2020年发布的《药品注册管理办法》标志着中国药品管理体制的全面升级,是药品领域重要的法规之一。这一办法的出台,不仅是对药品行业的严格要求,也是对制药企业发展创新的鼓励与支持。随着全球制药行业的不断变化和我国对药品监管要求的提高,新的管理办法在药品研发、注册、审批等多个环节都进行了调整和优化,目的是促进药品质量的提高,提升产业创新能力,保障公众用药安全。
2020药品注册管理办法提出了更加严格的药品注册要求。对药品的安全性、有效性和质量一致性提出了更高的要求,确保药品进入市场前经过严格的审查和评估。比如,对于药品临床试验的要求更加细化,确保临床试验数据真实、完整且具有代表性,避免在试验过程中出现偏差。与此新的办法在药品注册过程中强调药品的临床试验数据透明性,确保每一项试验结果都能够被及时公开和接受社会监督。
2020年药品注册管理办法加强了对药品生产质量的监管。办法要求药品注册的申请人必须提供药品生产过程中所有环节的详细信息,确保药品的生产符合GMP(良好生产规范)要求。这一要求对制药企业提出了更高的生产标准,企业不仅需要加强生产环节的质量管控,还需要在设备、人员、工艺等方面不断提升,以保证药品生产的一致性和稳定性。

2020药品注册管理办法也对药品创新进行了鼓励与支持。随着我国制药产业向高端化、创新化发展,新的办法在药品注册审批上给予了一定的灵活性。例如,办法对创新药品的注册审批流程进行了简化,缩短了药品的审批周期,为企业提供了更为在线画图工具的市场通道。办法还明确了针对稀缺病种、重大疾病药品的优先审评政策,对具有显著临床治疗效果的药品予以优先审批,以鼓励创新药品的研发,满足临床需求,推动国内制药企业在全球市场中的竞争力。
对于药品注册管理办法的具体实施,除了政策调整外,还有一些细节上的变化值得企业关注。2020年的新版办法明确了药品注册申请的时间要求和审批流程,使得企业能够更清楚地了解每一阶段的具体要求,避免因信息不对称或流程不明确而产生的困扰。例如,办法中规定,药品注册的审批流程分为初审、技术审查、综合评审等多个环节,每个环节都有明确的时间限制,确保审批过程高效有序。
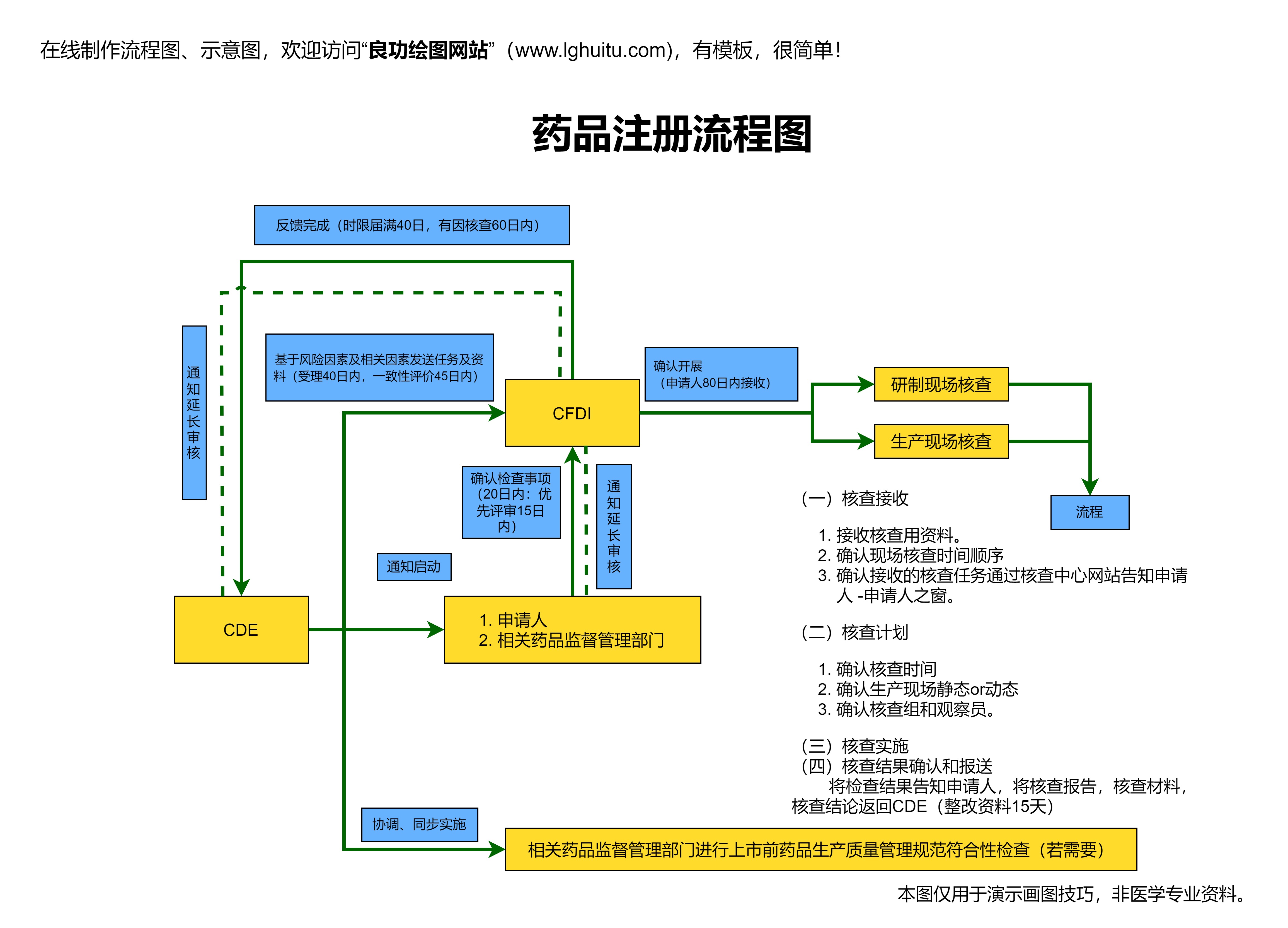
新办法还特别强调了对药品注册申请过程中所需资料的准确性和完整性。根据办法规定,企业在提交药品注册申请时必须提供所有必要的技术文件和试验数据,并且对这些数据的真实性负责。这一要求不仅提高了药品注册的透明度,还降低了审批过程中人为干扰的风险,确保了审批过程的公正性。
新的药品注册管理办法还在国际化方面做出了积极探索。为了促进中国制药行业的国际化发展,办法在审批过程中引入了国际标准的评审机制。具体而言,药品研发企业若能够在其他国家或地区获得药品批准,那么在我国的注册审批过程中可以申请加快审批。这不仅有助于我国制药企业更快进入国际市场,也提高了我国药品行业的全球竞争力。
随着药品注册管理办法的不断完善,药品行业的市场环境也发生了深刻变化。越来越多的药品研发企业开始将重点放在创新药物的研发上,药品的研发周期得到了大幅缩短。政策的优化为企业提供了更为广阔的创新空间,也使得企业在满足监管要求的能够更高效地推动新药的开发和上市。
2020年药品注册管理办法的出台,标志着中国药品管理进入了一个新阶段。这一办法不仅强化了药品注册的监管力度,还为药品创新提供了更加宽松的政策环境。对于制药企业来说,如何适应这一新规定,并通过创新提升产品竞争力,将是未来发展的关键。而对于广大患者而言,新的管理办法必将进一步保障他们的用药安全,带来更多高效、安全、创新的药物选择。