在当今快速发展的制药行业中,药品的注册审批流程至关重要,它不仅关系到药品能否顺利上市,还直接影响到制药企业的竞争力与市场份额。而《药品注册管理办法》作为药品注册的重要法律法规文件,其规范的药品注册流程,不仅有助于提升药品的质量安全,还为药品企业提供了明确的操作指引。本文将通过一张清晰的流程图,详细解析《药品注册管理办法》的核心流程,帮助企业和行业从业者更好地理解药品注册的每个关键环节。

我们需要明确《药品注册管理办法》的总体框架。这一办法规定了药品从申请注册到最终获得批准上市的全过程,主要包括药品注册申请的受理、审评、审批以及批准后的跟踪管理等环节。通过合理的流程管理,能够确保每一款药品都经过严格的审查与评估,从而保证药品的安全性、有效性和质量可控性。
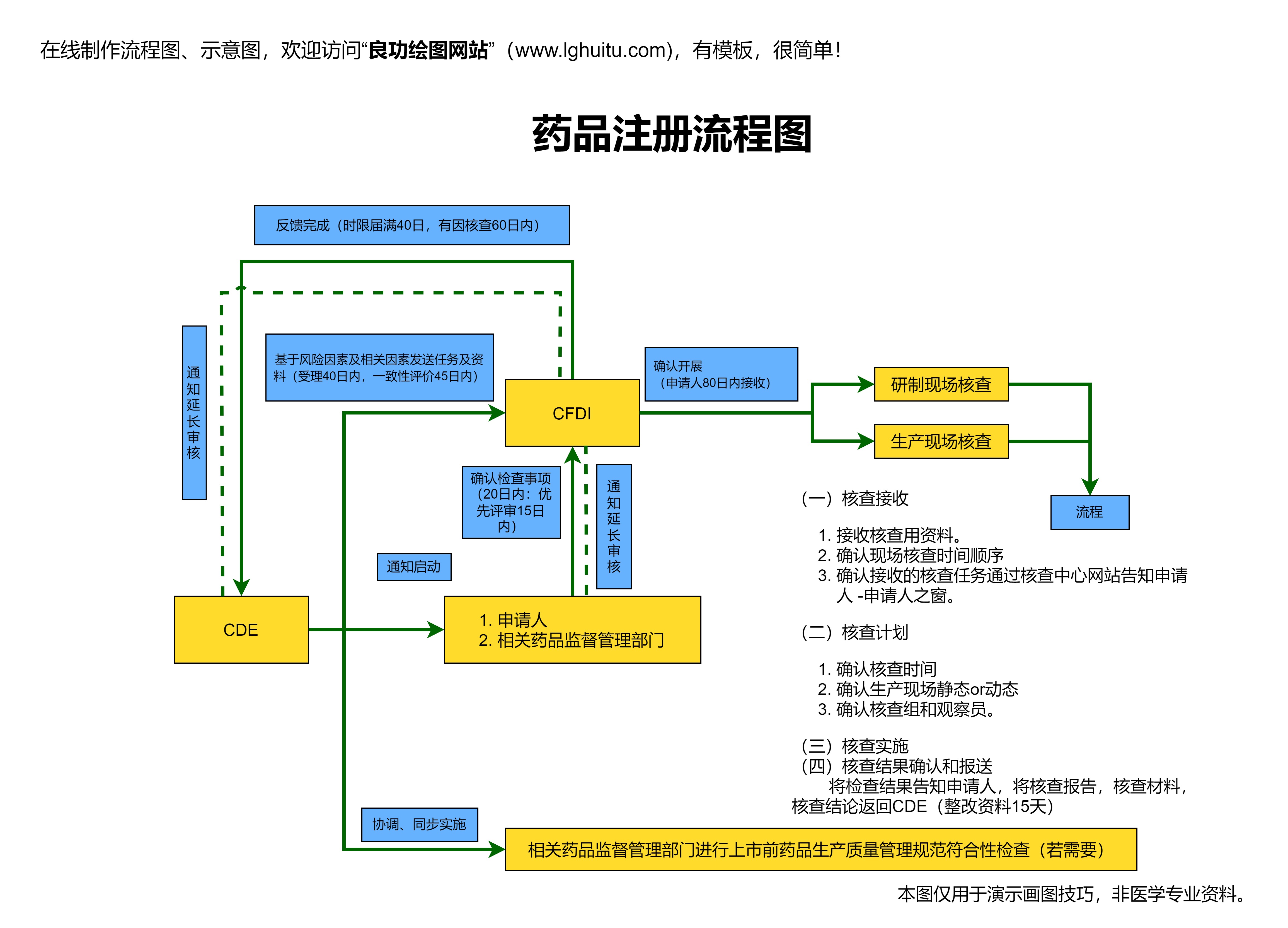
根据《药品注册管理办法》的要求,药品注册的流程大致可以分为以下几个阶段:首先是申请人提交注册申请,接下来是药品注册受理阶段;然后是药品的技术审评阶段,再到药品的审批阶段;最后是药品的生产、流通和上市后监督管理。每一个环节都有严格的规定和要求,任何一个环节的疏忽,都可能导致注册过程的延误或失败。
在药品注册的第一步,企业需要向国家药品监督管理局(NMPA)提交药品注册申请。这个过程非常重要,要求企业提供完整的药品注册资料,包括药品的成分、剂型、生产工艺、临床试验数据等。这些资料的完整性和准确性直接决定了药品注册的顺利程度。因此,企业在提交资料时一定要确保所有信息的真实性和完整性。
药品注册的受理阶段将决定是否进入下一步的审评程序。在这一阶段,NMPA会对药品注册资料进行初步审核,确认申请资料是否齐全和符合相关法规。如果资料存在缺漏或不符合要求,企业需要按要求进行补充和修改。只有当资料完整无误时,药品才会正式进入技术审评阶段。
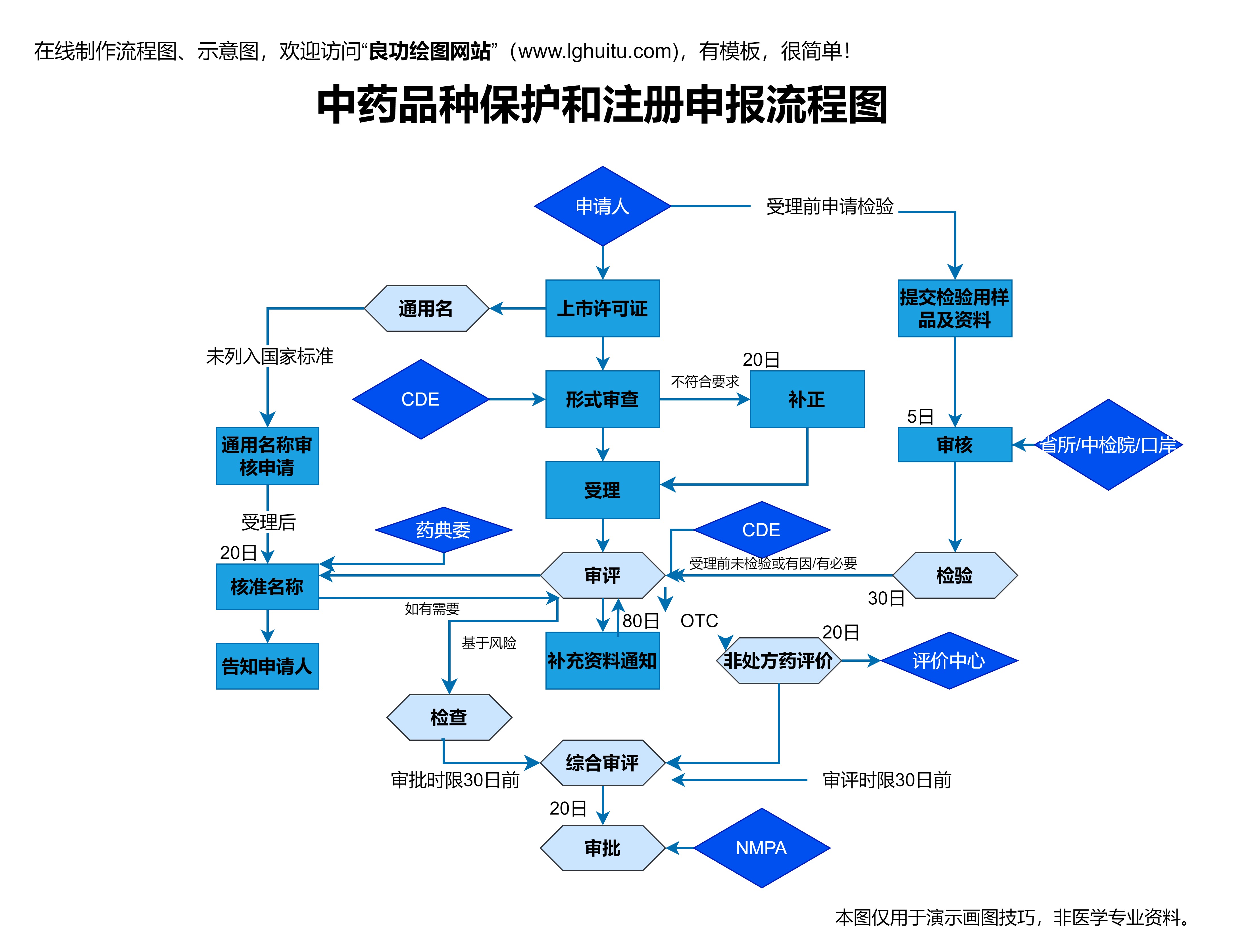
技术审评阶段是药品注册过程中的核心部分,NMPA将对药品的临床试验数据、药品质量控制标准、生产工艺等内容进行严格审查。这一阶段是药品是否能够顺利获得批准的关键。如果药品在此阶段未能通过审评,将被要求进行整改,甚至可能面临驳回申请的风险。因此,企业在研发药品时,必须确保其产品符合国家药品管理法规的相关要求。
技术审评通过后,药品注册将进入审批阶段。审批阶段是药品注册的最后环节,一旦通过审批,药品即可获得上市批准,并进入市场流通。这一阶段虽然看似是药品注册的终点,但实际并非如此,药品在上市后还需要接受持续的监管。上市后监管的内容包括药品的生产、质量控制以及不良反应的监测等。
《药品注册管理办法》在确保药品上市后的安全性方面,明确了药品不良反应的监测机制。企业需要建立健全的药品不良反应报告制度,并及时向相关部门报告。通过对药品的不良反应进行跟踪和管理,能够进一步提升药品的安全性,避免因药品的不良反应造成公众健康问题。
药品的生产和流通环节也受到严格的监管。只有当药品生产企业通过了GMP认证,确保其生产过程符合严格的质量标准,药品才能进入市场。药品的流通环节也需要符合国家药品管理法规,确保药品的质量在流通过程中不受污染或损坏。
为了帮助企业更好地理解药品注册过程,下面我们提供了一张详细的《药品注册管理办法》流程图。通过流程图,您可以清晰地看到药品注册的各个环节及其间的关系。这张流程图不仅能够帮助企业在实际操作中理清思路,避免遗漏关键步骤,还能有效提升药品注册的工作效率。
《药品注册管理办法》不仅是药品注册过程的法律依据,也是企业合规经营、提升市场竞争力的重要指南。随着制药行业的不断发展和药品注册要求的不断提高,企业在进行药品注册时必须严格按照《药品注册管理办法》的要求,确保每一个环节都做到合规和精细化操作。通过对流程图的解析和对每一环节的细致讲解,企业将能够更好地掌握药品注册的要点,推动药品的顺利上市,并为患者带来更安全、更有效的药品。