随着医学科学的不断进步,一类新药的研发和审批已成为全球医药产业中的重要环节。从一个药物的研发立项到最终上市,每一项步骤都需要经过严格的审查和评估,确保新药的安全性、有效性及质量可控。这不仅是对患者健康的承诺,也是对医药产业质量控制的高度要求。一类新药的审批流程究竟是怎样的呢?本文将从多个角度为您详细解析。
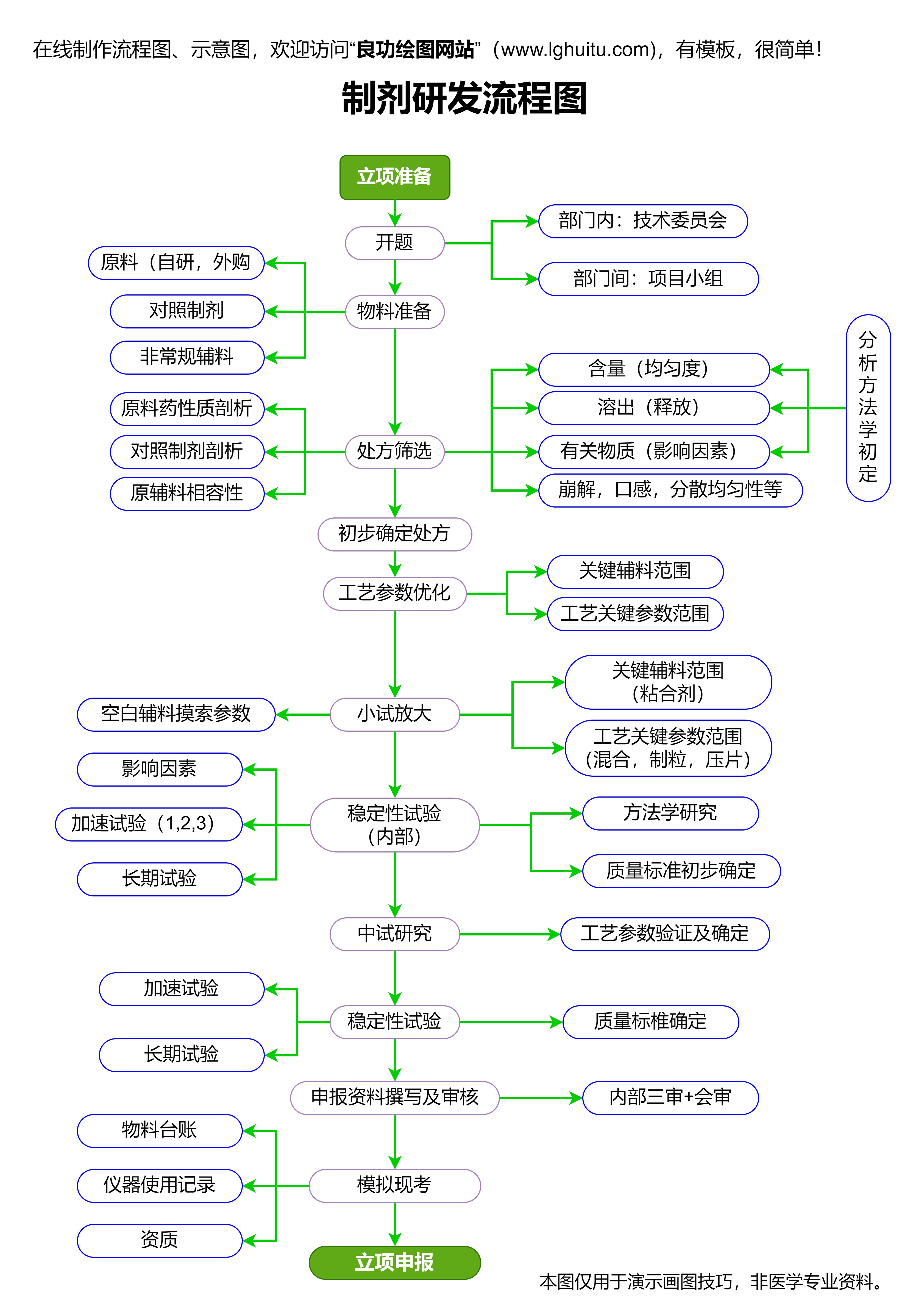
我们需要了解一类新药的定义。一般而言,一类新药是指在全球范围内首次被发现、首次研发的药物,具有全新的作用机制或疗效,并未曾在国内外药品市场上上市。因此,通常情况下,这些药物在研发阶段涉及到前所未有的挑战。对于研发企业而言,能够研发出一类新药不仅是一项巨大的技术突破,也是市场竞争力的重要体现。
开发一类新药的过程是漫长且复杂的,涉及到从初步的化学结构研究到临床数据分析等多个环节。而其中的审批流程更是每一位药品研发人员不可忽视的关键步骤。
药品的研发分为多个阶段,通常包括发现阶段、早期研发阶段、临床试验阶段和商业化阶段。在药品发现阶段,研究人员会通过对疾病机制的研究,找出潜在的治疗靶点。这个阶段的核心目标是筛选出有可能具有临床应用价值的候选分子。药物进入早期研发阶段,通常会涉及药理学、毒理学、制剂学等多个学科的研究。
一旦研发人员确认候选药物具有治疗潜力,并通过了前期的安全性和效果测试,药物就进入了临床试验阶段。临床试验是整个药物研发过程中的核心部分,也是药品能否上市的关键。临床试验分为I期、II期、III期三个阶段,每个阶段的目标不同,从小范围的安全性测试到大规模的疗效验证,逐步积累数据,为后续的药品批准提供科学依据。
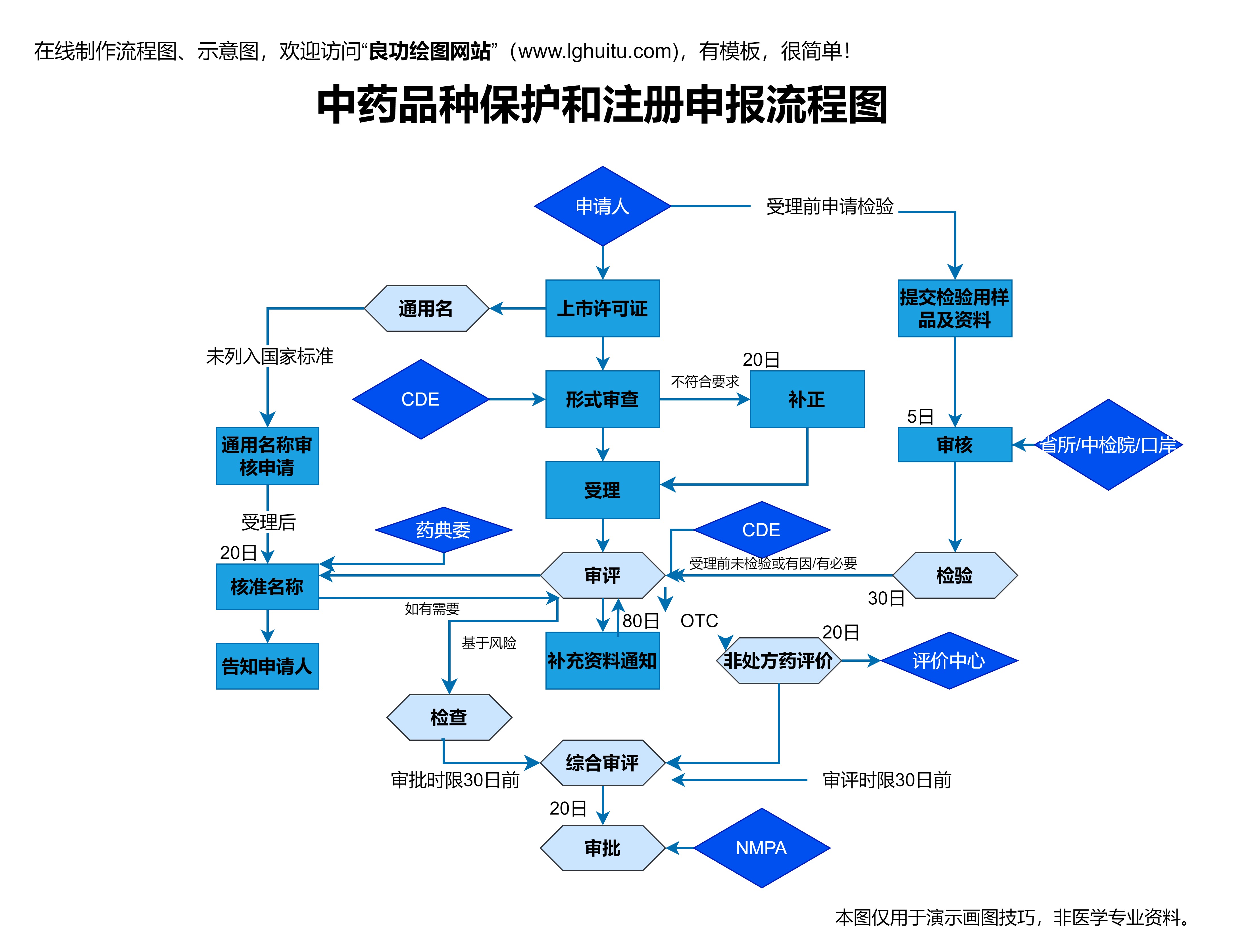
药品研发完成后,企业需要向国家药品监管部门提交新药注册申请,申请包括药物的临床试验数据、生产工艺、药品质量控制标准等详细信息。这一环节需要进行严格的审核,确保药物的质量标准符合国家要求。
药品注册的审批流程较为复杂,涉及多个环节的评审。企业需要向药品管理局提交临床前数据,包括毒理学、药理学等研究成果。还需要提供大量的临床数据,证明该药物在治疗某种疾病中的疗效。审批部门会基于这些数据,进行严格评估。

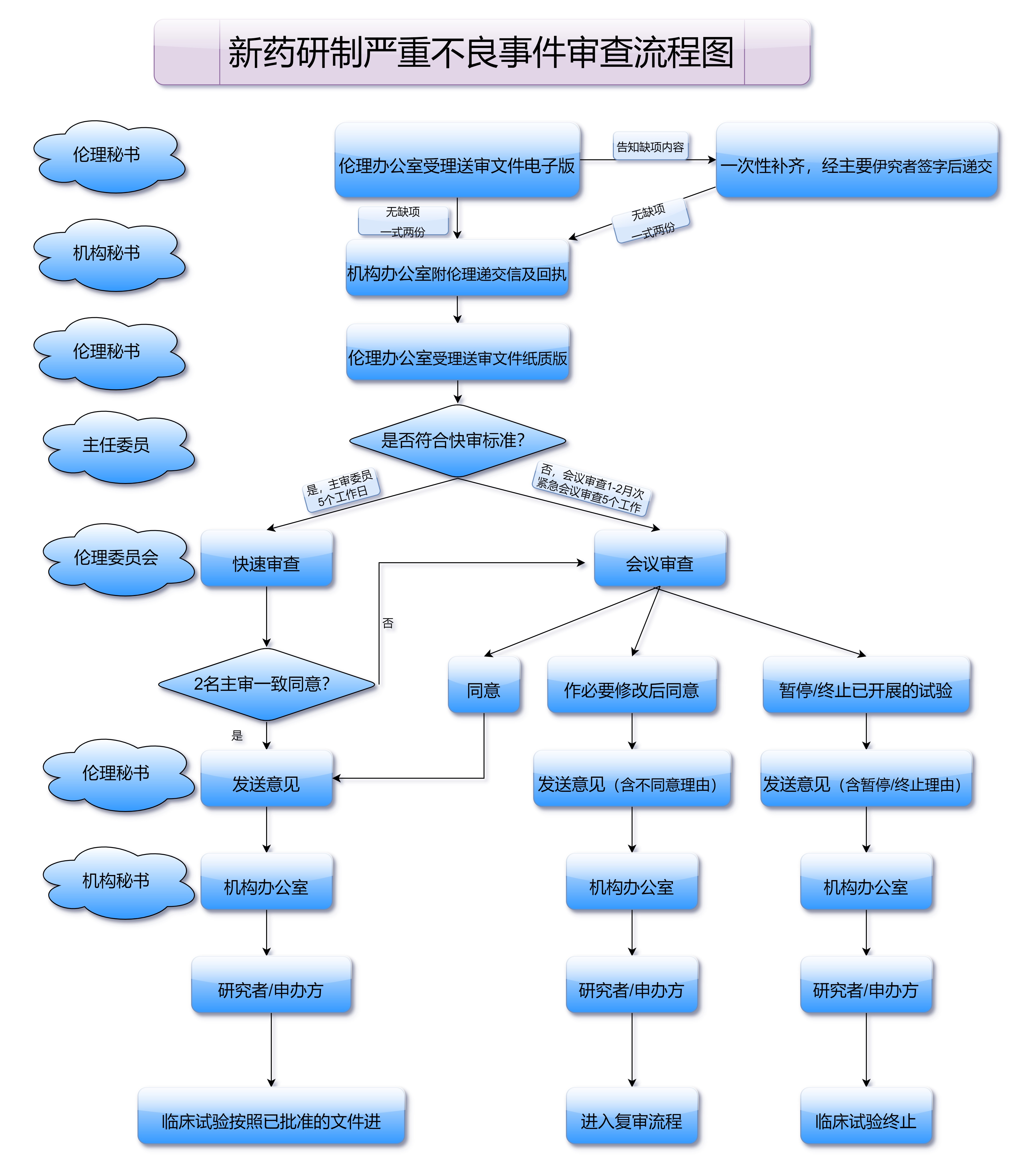
在临床试验过程中,药品需经过药监局的批准才能开始。药品的临床试验需要向监管机构提交详细的试验方案,包括试验的设计、样本量的计算、药物的给药方式等内容。临床试验的审批环节不仅需要药品研发企业的详细资料支持,还需要通过伦理委员会的审查,以确保患者在试验过程中的安全性。
一旦临床试验获批并开始实施,药品研发团队将会根据试验结果逐步完善药物的临床应用数据。I期临床试验主要针对药物的安全性进行评估,测试药物在人体中的耐受性。II期临床试验则关注药物的初步疗效,同时进一步评估药物的安全性。而III期临床试验则是进行大规模患者群体的测试,确认药物的有效性和长期使用的安全性。
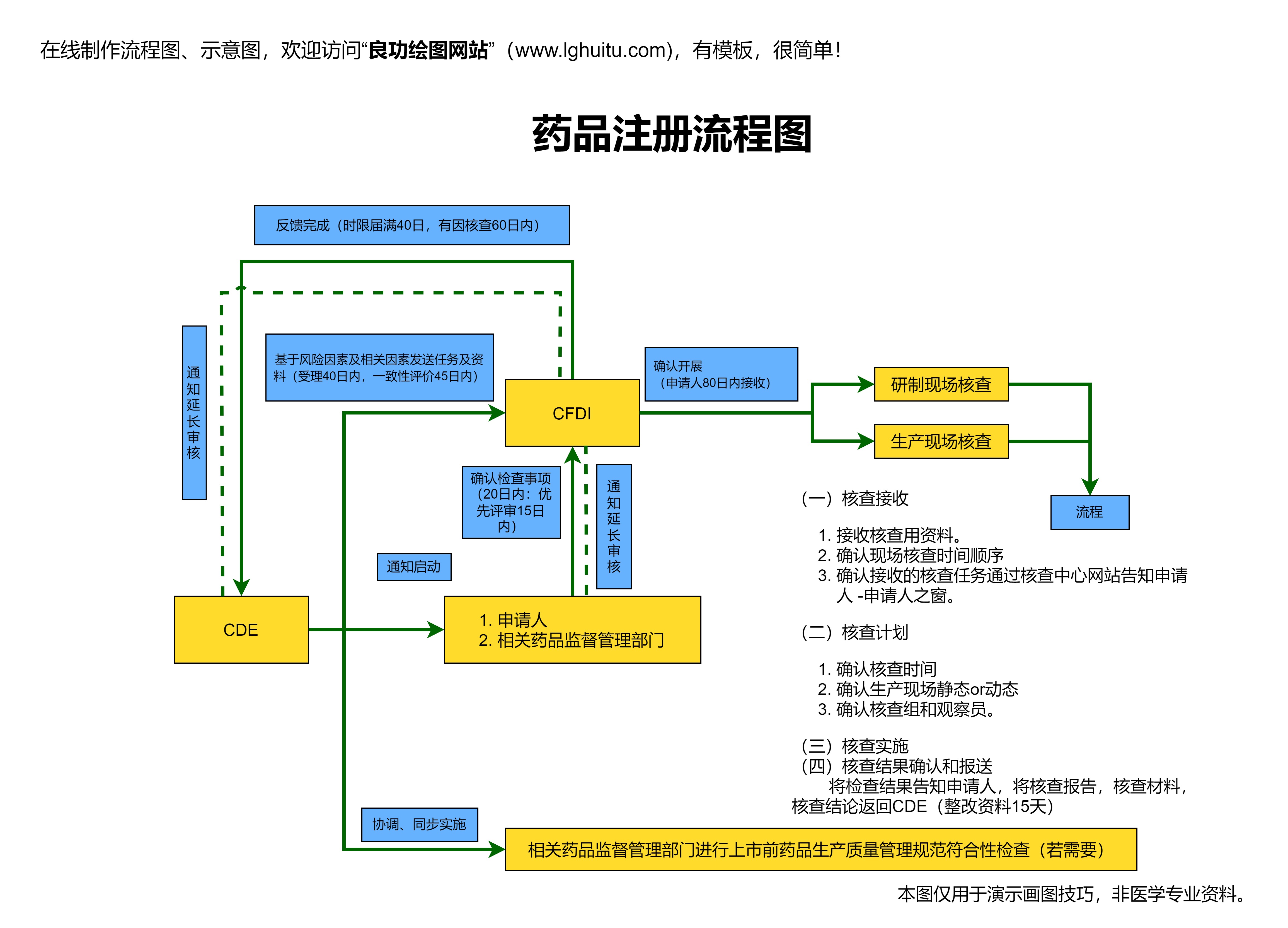
完成临床试验后,研发企业可以向药品监管部门提交“上市申请”,也就是新药申请的关键步骤。在这一阶段,监管部门将会对药物的所有数据进行审查,确保药物的质量、安全性以及临床效果符合上市要求。药品的质量控制要求严格,尤其是生产工艺的稳定性、成品的质量控制等内容都需要经过严格验证。
药品注册申请一旦通过,药品将获得上市许可,并可以在市场上销售。值得注意的是,在药品上市后,药品仍需接受监管部门的持续监控。药品上市后的不良反应监测、临床效用跟踪等工作,也是确保药品长期安全使用的重要保障。
对于药品研发公司来说,一类新药的审批不仅是技术和资本的考验,更是创新和市场机遇的争夺。在全球范围内,新药的研发和审批周期长、成本高,尤其是面对国内外法规政策的差异,药品研发企业面临着巨大的压力。这也是一次证明企业创新能力和技术实力的重要机会。
随着国内药品审批制度的逐步完善,中国药品监管部门对新药审批流程的透明度和高效性不断提升。在符合国际标准的审批流程下,越来越多的创新药物能够进入市场,为患者带来更多的治疗选择。国家对药品研发的政策支持也在不断加大,从税收优惠到资金补贴,各项政策措施为新药研发提供了有力的支持。
一类新药的审批流程不仅是一项复杂的技术性工作,更是保障患者健康、推动医药科技进步的关键环节。对于药品研发企业来说,每一个环节都充满了挑战,而每一次突破也都是创新与责任的体现。在未来,随着科技的发展和审批流程的不断优化,我们有理由相信,更多具有突破性意义的新药将进入市场,为全球患者带来福音。