在全球制药行业中,一类新药的注册申报无疑是一个复杂且挑战性的过程。从药品的临床研究到注册资料的提交,每一步都需要严格遵循法规要求,并且具有极高的专业性。对于药品研发公司来说,了解并掌握一类新药的注册申报流程,不仅是顺利进入市场的关键,更是确保药品质量和安全的前提。本文将深入解析一类新药注册申报的详细流程,帮助您一步步完成注册,实现商业化成功。
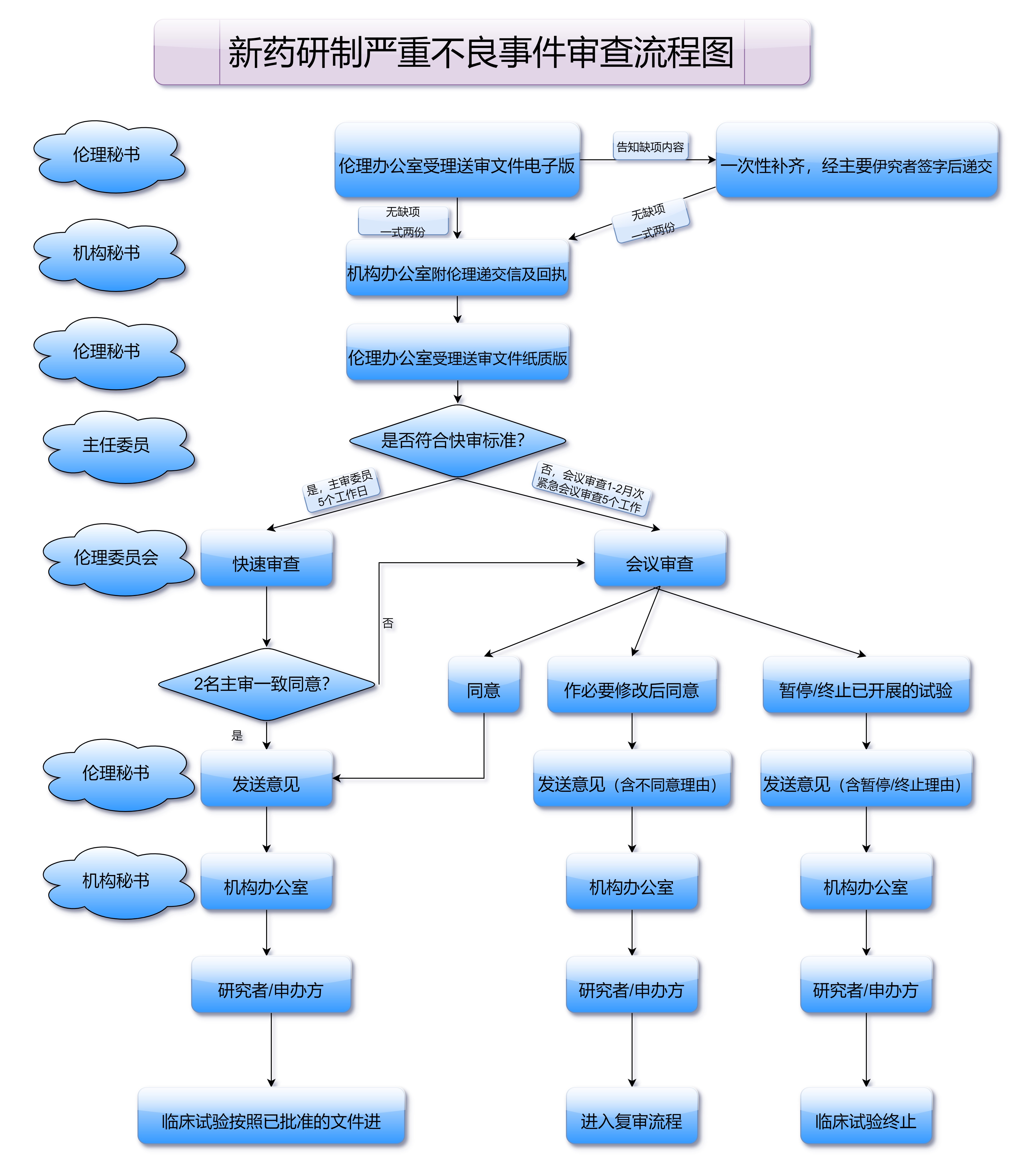
在正式进行一类新药注册申报之前,药品的研发和临床试验是至关重要的前期准备工作。药品的有效性与安全性必须通过临床前研究得以确认。临床前研究包括药物的药理毒理、药代动力学等一系列实验,这些研究结果将作为申请药品注册的基础资料之一。

随后,药品进入临床试验阶段。中国药品管理法要求新药必须进行严格的三期临床试验,来评估药物在人群中的安全性和有效性。第一期临床试验主要用于评估药品的安全性;第二期试验评估药物的有效性和安全性;而第三期临床试验则进一步验证药物在大规模人群中的效果和安全性。整个过程需要大量的时间和资金投入,同时也要时刻遵守国家药品管理相关规定。
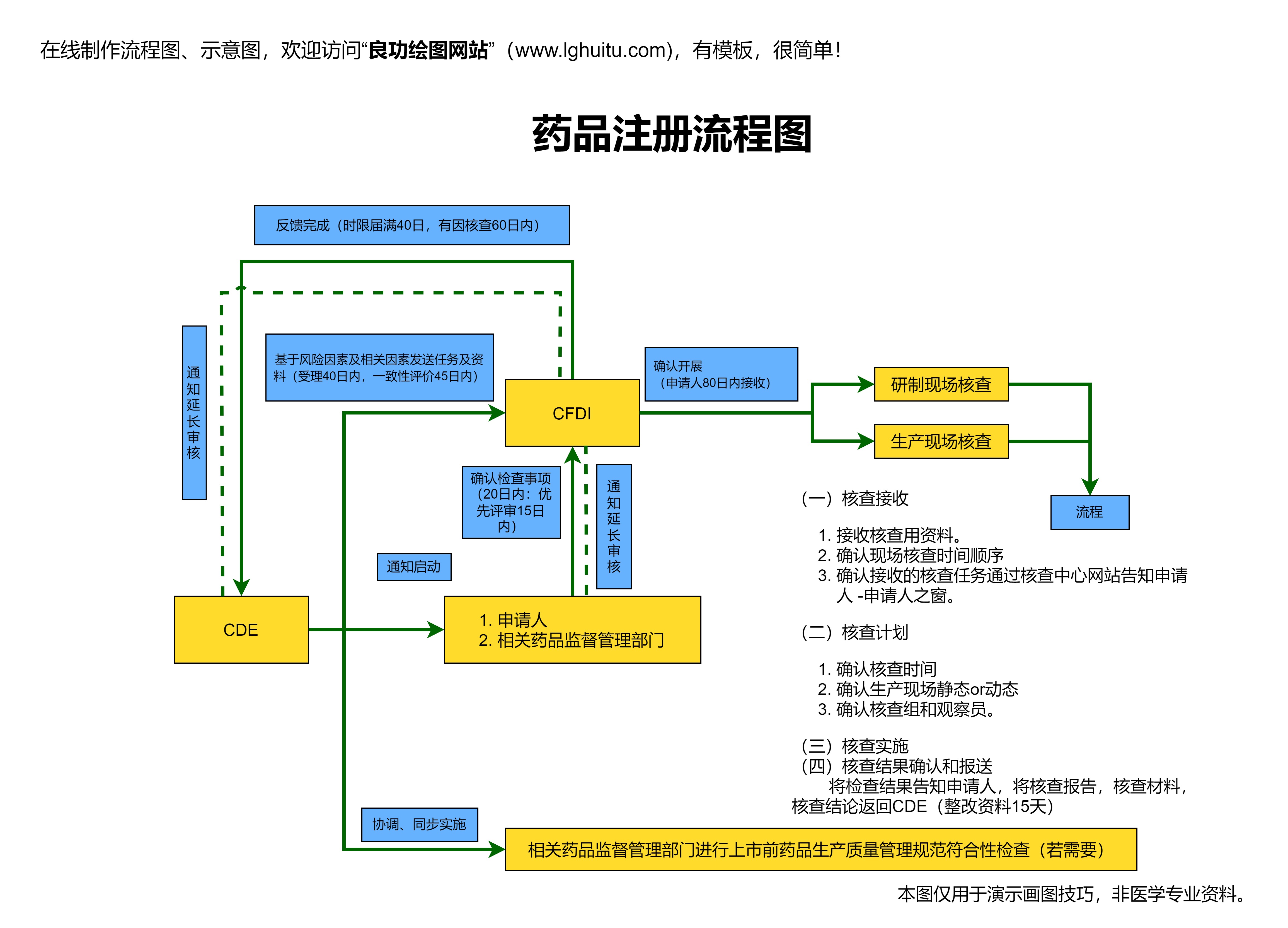
当临床试验阶段完成后,药企可以准备开始注册申报工作。首先要向国家药监局(NMPA)提交药品注册申请。在这个阶段,药企需要准备详尽的资料,包括药品的研发背景、临床试验数据、生产工艺、药品质量标准等。具体来说,注册申报材料大致包括以下几个方面:
药品基本信息:包括药品的名称、规格、剂型、适应症等基本信息。
临床试验数据:包括临床试验的全部数据报告,必须详细展示药品的有效性、安全性及其治疗优势。
生产工艺和质量标准:药品的生产过程必须符合国家药品生产质量管理规范(GMP)的要求,确保药品的质量与安全。
药品标签和说明书:药品标签与说明书必须清晰明确地说明药品的使用方法、副作用、禁忌症等关键信息。
准备这些资料时,药企需要特别注意资料的完整性和规范性,任何细节问题都可能影响注册的进度与成功率。
药品注册申请的核心是向国家药监局提交一份完整的药品注册申报材料。材料提交后,药监局会对提交的资料进行详细审评,审核的内容包括药品的临床数据、生产工艺、质量标准以及药品的安全性和有效性等。在此过程中,药监局可能会要求药企提供补充资料或进行进一步的临床研究,以确保药品符合国家相关法规和标准。
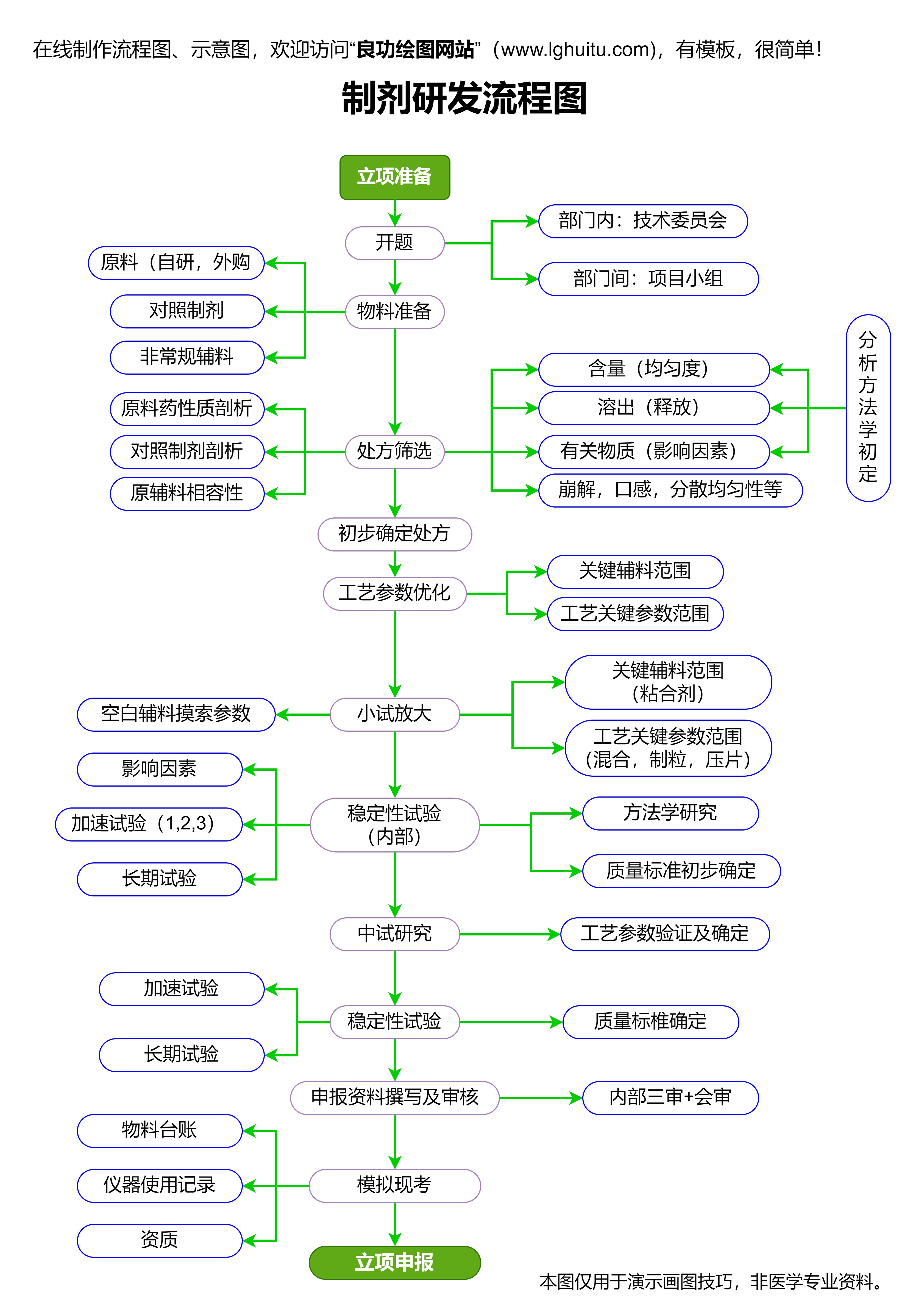
药品审评通常分为几个环节:初审、技术审评、专家评审等。审评工作通常需要一定时间,且药企需要与药监局保持良好的沟通,以便及时了解审评进度并提供必要的支持。
当药品的注册申请通过审评后,药监局将会发放《药品注册批件》。这个批件意味着药品在中国市场的正式获批,药企可以开始在国内市场销售该药品。但在正式销售之前,药企还需要获得药品的生产许可证。
获得药品生产许可证的前提是药品的生产工艺符合国家药品生产质量管理规范(GMP)的要求。为了确保药品的质量,药企需要通过药监局的生产现场检查,确保所有生产环节符合规定,并且能够生产出符合标准的药品。一旦通过检查,药企就可以获得药品的生产许可证。
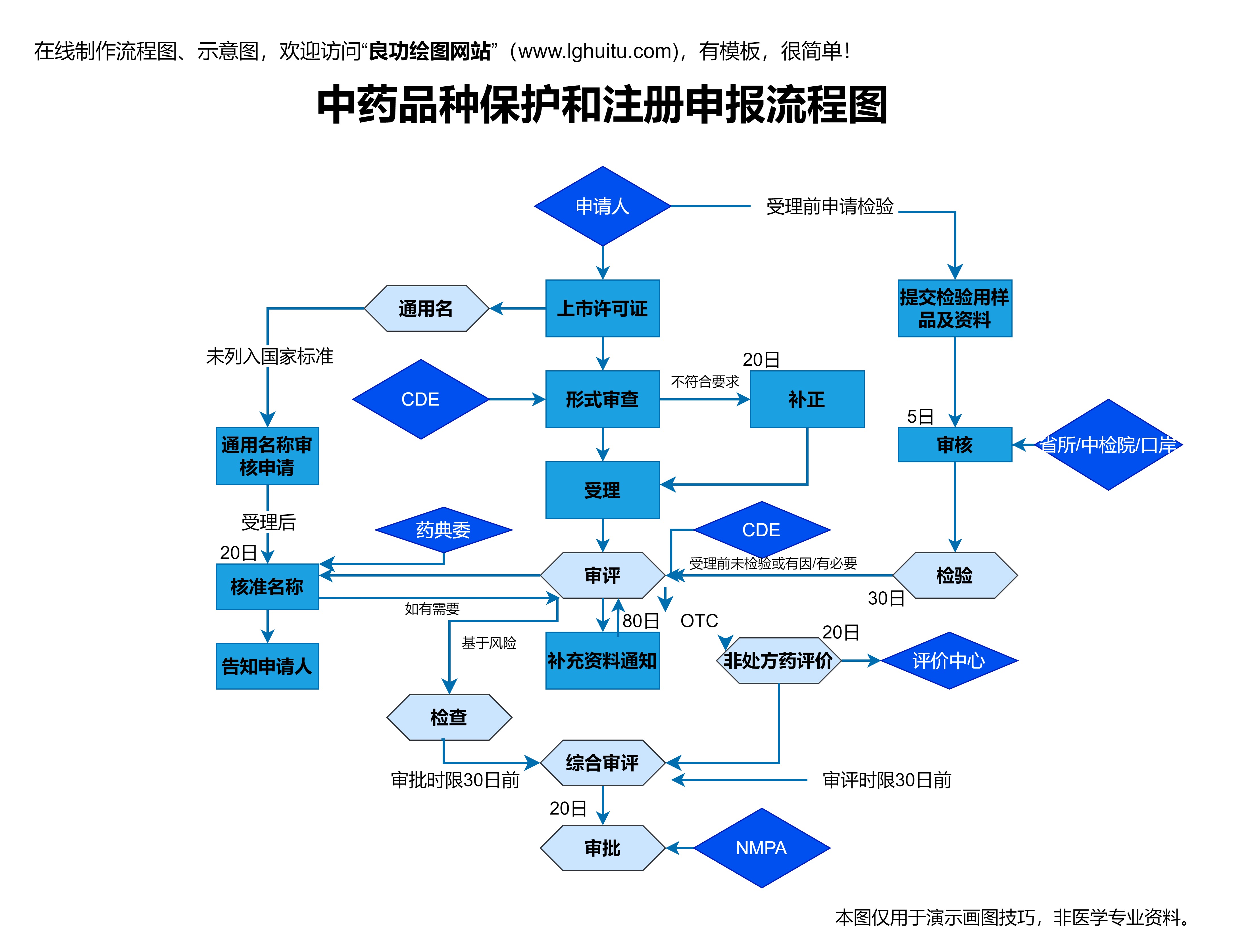
虽然一类新药的注册工作在获得药品注册批件后就可以结束,但药品的上市并不意味着监管的终结。药品上市后,药监局会持续进行药品的监管,包括药品的市场抽查、药品的不良反应报告等。药企在药品上市后,必须定期向药监局报告药品的安全性与有效性数据,并及时处理药品的不良反应和投诉。
药品的生产、销售、广告宣传等活动也需要遵循国家相关法规的要求,任何违法违规的行为都可能导致药品的撤市或者处罚。因此,药企必须时刻关注药品的生命周期管理,确保药品在市场上的持续合规性。
精准的市场定位与研发策略:一类新药的注册申报不仅需要扎实的科研基础,还需要在市场上找准定位。研发过程中,药企需要根据临床需求和市场情况进行科学合理的定位,确保药品能够满足特定人群的需求。
严格的质量控制与生产管理:无论是临床试验还是药品生产,质量控制都是重中之重。药企必须严格执行GMP标准,确保药品质量过硬,从源头上杜绝可能的质量问题。
与监管部门的良好沟通:在药品注册过程中,药企需要与国家药监局保持密切的沟通,及时了解政策变化和审评进度,以便做出快速反应。
总结而言,一类新药的注册申报是一个系统而复杂的过程,涉及到从研发、临床试验到申报、审评等多个环节。只有通过精心的准备、严谨的执行和与监管部门的密切合作,药企才能够成功获得药品注册批件,实现新药的顺利上市,为患者提供创新的治疗选择。