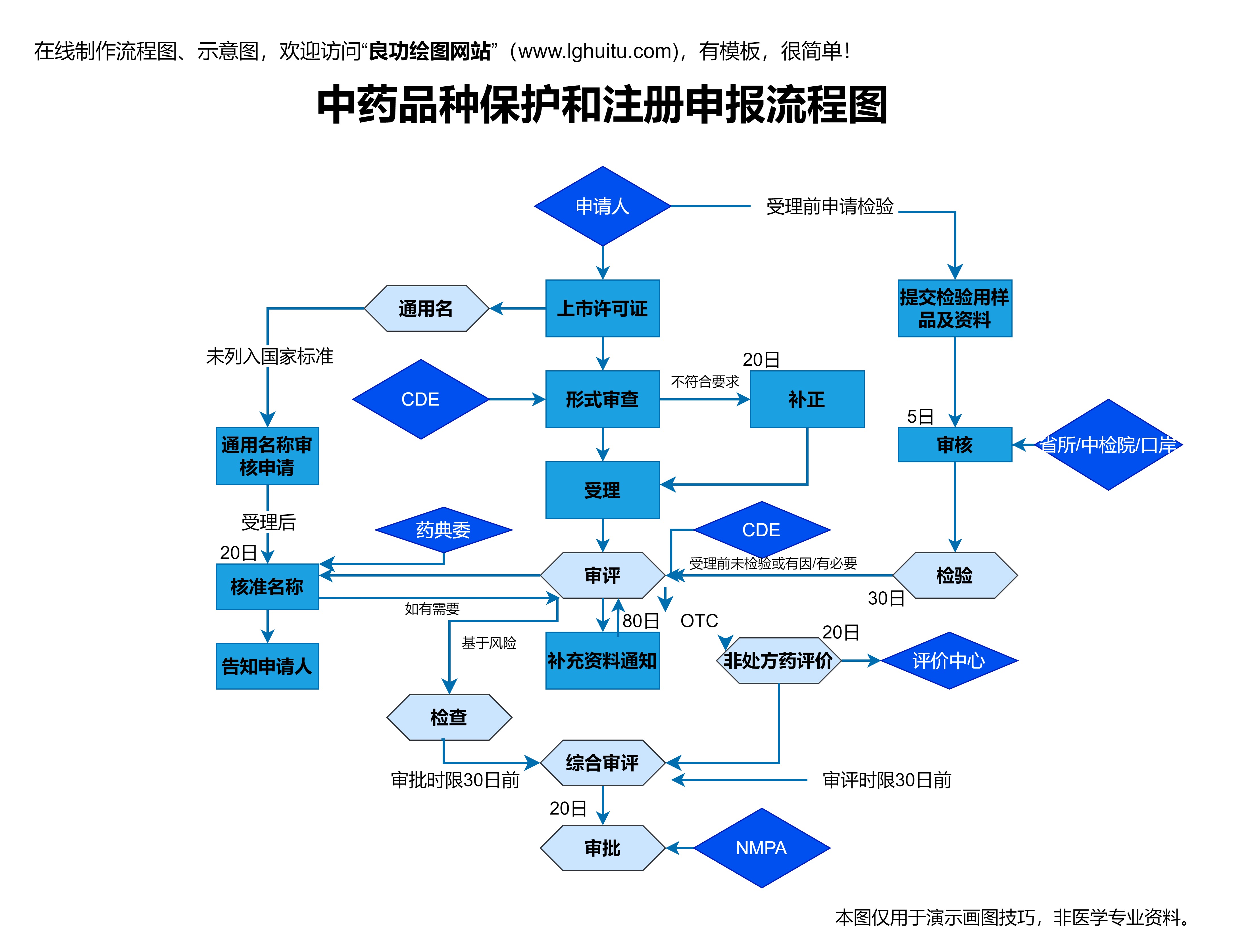
在中国,随着人们对健康生活的重视和中医药文化的复兴,中成药的市场需求日益增长。为了确保中成药的质量和疗效,每一款中成药都需要经过严格的注册程序,方能合法上市。作为企业或制药商,了解和掌握中成药注册流程至关重要。本文将详细解析中成药注册的关键环节,帮助您顺利通过注册关卡,让您的产品走向市场。
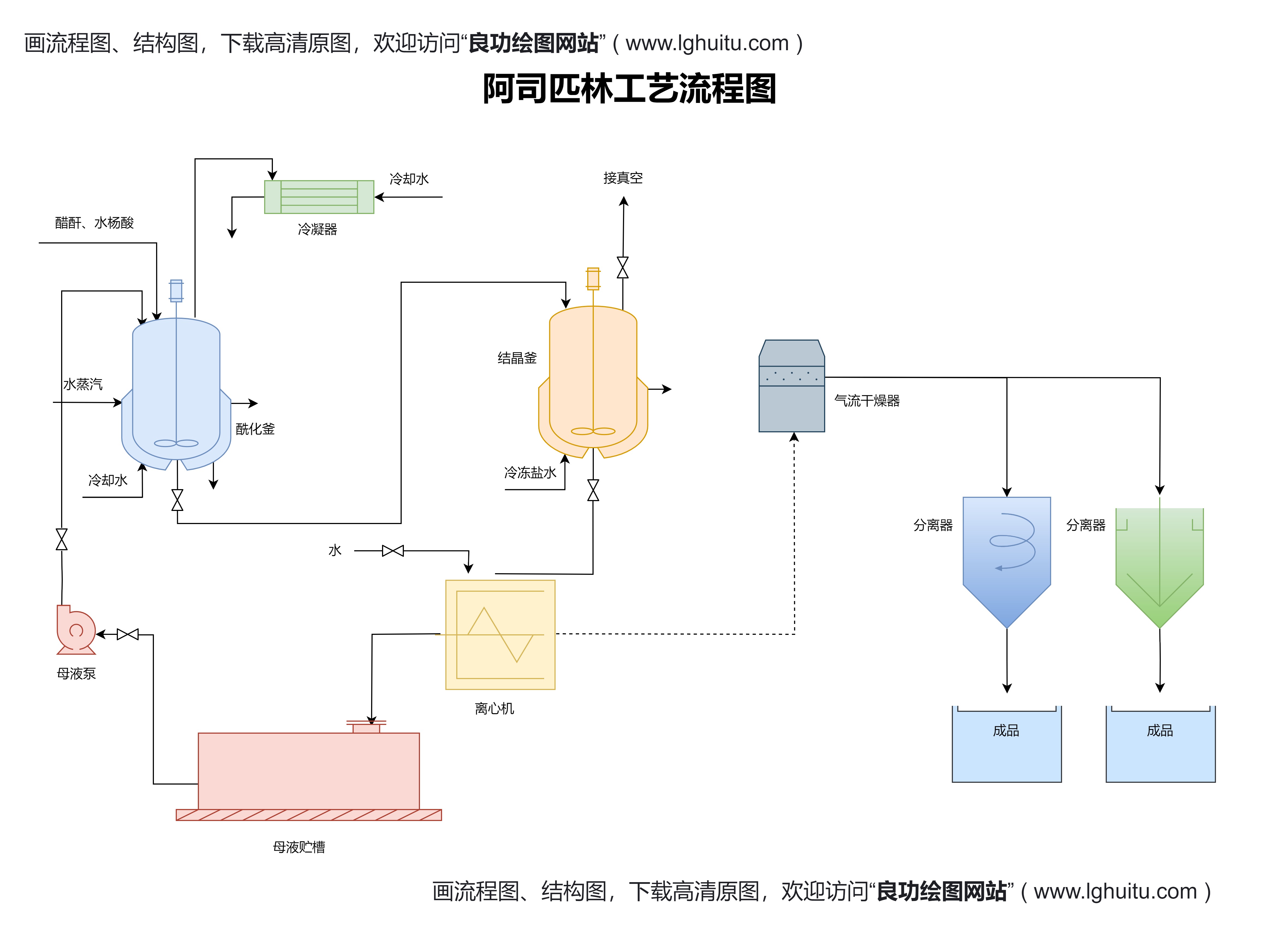
中成药的注册管理涉及到国家药品监督管理局(NMPA)以及其他相关法律法规的规定。中成药作为一种特殊的药品,必须符合《药品管理法》、《中药管理法》和《药品注册管理办法》等法律要求。对于企业而言,必须在符合药品注册条件的基础上进行相应的申报工作,确保产品的安全性、有效性和质量可控。
在中国,进行中成药注册时,企业需要准备充分的材料,包括药品的成分、生产工艺、质量标准以及临床试验等资料。一般而言,中成药的注册流程可以分为两个大类:新药注册和仿制药注册。新药注册的要求相对严格,通常涉及到从药理研究到临床试验的全过程,而仿制药注册则侧重于产品与原研药的相似性和质量控制。

在正式提交注册申请之前,企业需要做好充分的准备工作。首先要确认中成药的产品类别,并根据药品的类型确定是否需要进行临床试验。如果药品属于创新性中成药,企业需要进行大量的实验室研究和动物实验,以证明其安全性和有效性。如果是仿制药,则需要提供原研药的相关资料,并进行一致性评价。
企业还需要与药品生产厂商密切合作,确保药品生产工艺的符合国家标准,并且能够保证产品的一致性和质量稳定性。药品的包装、标签、说明书等也需要符合国家相关规定。
在准备工作完成后,企业可以开始进行注册申请。根据药品注册管理的相关规定,申请人需要向NMPA提交一系列材料。这些材料主要包括:
药品的研究资料:包括药品的成分、作用机制、质量标准、生产工艺等;
临床试验资料(如适用):证明药品在人体中的安全性和有效性;
提交的材料需要通过NMPA的初步审查,确保其符合药品注册的基本要求。
一旦注册申请材料提交到NMPA,药品注册进入了审评阶段。NMPA会对所有提交的资料进行详细审查,确保药品的安全性、有效性和质量可控。审评过程通常会根据药品的不同性质有所不同。如果是创新药,审评时间可能较长,需要进行更加严格的临床试验和数据分析。
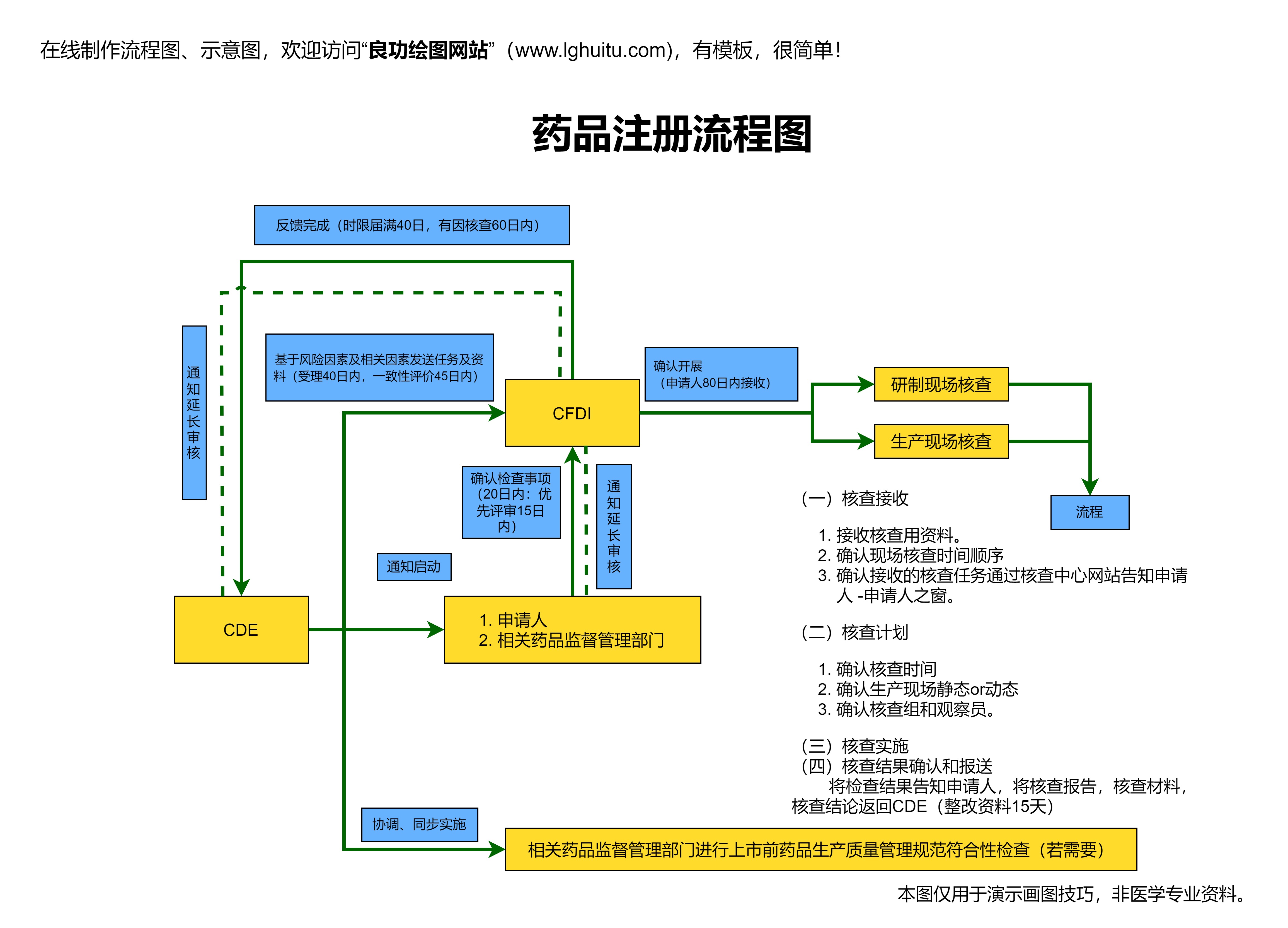
对于中成药的注册,尤其需要注意的是,药品的质量标准和生产工艺必须符合中国药典的相关要求。如果药品已经在其他国家或地区上市,NMPA也会参考国际认证和审核资料,以加速审评过程。
对于部分中成药,尤其是新药,临床试验是注册过程中的重要环节。企业需要按照国家药监局的要求,开展相应的临床前研究以及临床试验。临床试验一般分为三个阶段:
I期临床试验:主要评估药物在人体中的安全性和剂量反应;
III期临床试验:进一步验证药物的疗效和安全性,为上市申请提供数据支持。
完成临床试验后,企业还需提交临床试验的总结报告,证明药品的临床效果和安全性。
即使中成药成功注册并上市后,企业仍然需要承担产品上市后的监管责任。根据《药品管理法》和相关法规,企业必须对药品的生产和销售过程进行严格的质量控制,并定期向药监部门报告产品的安全性和有效性。若发生药品不良反应,企业需要及时报告,并采取相应的处理措施。
中成药的注册是一个复杂而严谨的过程,涉及到从药品研究到临床试验,再到产品生产和上市后的监管等多个环节。企业需要投入大量的时间、资金和人力资源,确保每一个环节都符合法律法规的要求,才能顺利将中成药推向市场。对于制药企业来说,了解整个注册流程并做好充分准备,是成功上市的关键。