在中国,药品的市场准入需要经过严格的审批过程,其中最重要的一项便是国药准字号。无论是国内企业还是外资企业,想要在中国销售药品,都必须通过国家药品监督管理局(NMPA)审批,并取得“国药准字号”认证。这不仅是药品合法上市的标志,也是产品质量和安全性的象征。如何才能顺利通过国药准字号的申请流程呢?本文将带您一步步了解这一过程的每个环节,帮助您顺利进入中国庞大的药品市场。
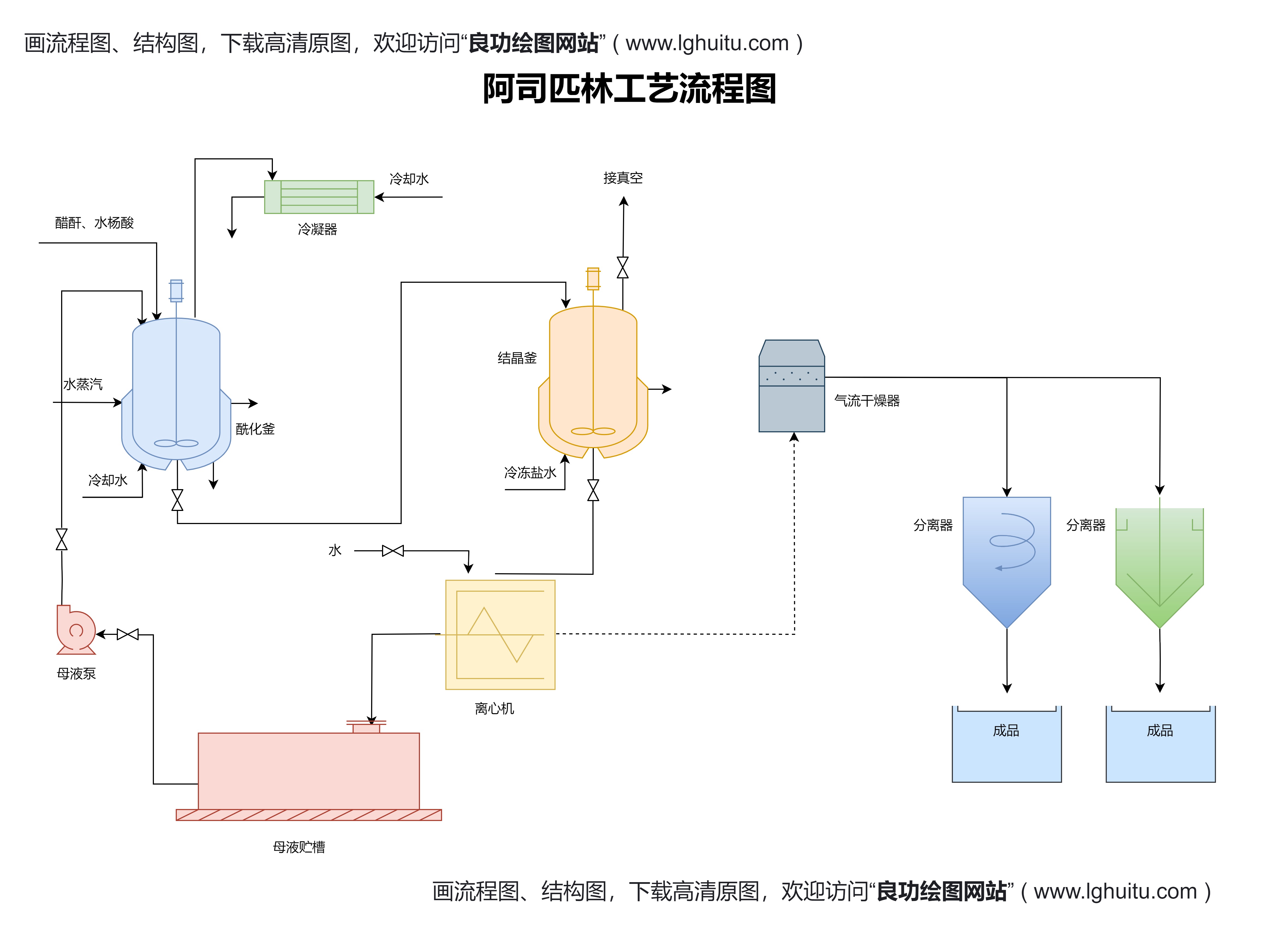
“国药准字号”是中国国家药品监督管理局(NMPA)授予合格药品的唯一标识,证明该药品经过严格的质量、安全和有效性评估,可以在中国市场合法销售。药品获得“国药准字号”后,意味着它已经符合中国药品管理法规的要求,可以进入各大医院、药店等销售渠道。
对于制药企业来说,国药准字号不仅是一项必要的法律要求,更是药品能否顺利销售的关键。如果您的药品未通过认证,则无法在中国市场上销售,因此,申请国药准字号显得尤为重要。
在开始申请流程之前,首先需要了解申请国药准字号所需满足的基本条件。通常,申请者需要具备以下条件:
药品的研究和生产符合规定要求:药品的研发过程必须符合中国药品法规的要求,生产设施需要达到GMP(药品生产质量管理规范)标准。
药品的临床数据和试验符合中国药品要求:无论是进口药品还是国内新药,都需要提交符合中国法规要求的临床试验数据。这些数据需证明药品的安全性、有效性以及质量可控性。
符合相关药品类别的管理规定:不同种类的药品(如化学药品、中成药、疫苗、诊断试剂等)所需的材料和审批要求不同。申请者需要根据药品的类型准备相应的资料。
注册申请人符合规定要求:注册申请人通常是药品的生产厂家,必须是依法注册并具有相关资质的企业。
申请国药准字号的第一步是进行前期准备工作。企业需要收集药品研发、生产、临床试验等方面的所有资料,并进行整理。这个阶段的核心工作是确保所有资料符合中国药品监管的标准和要求。具体包括:
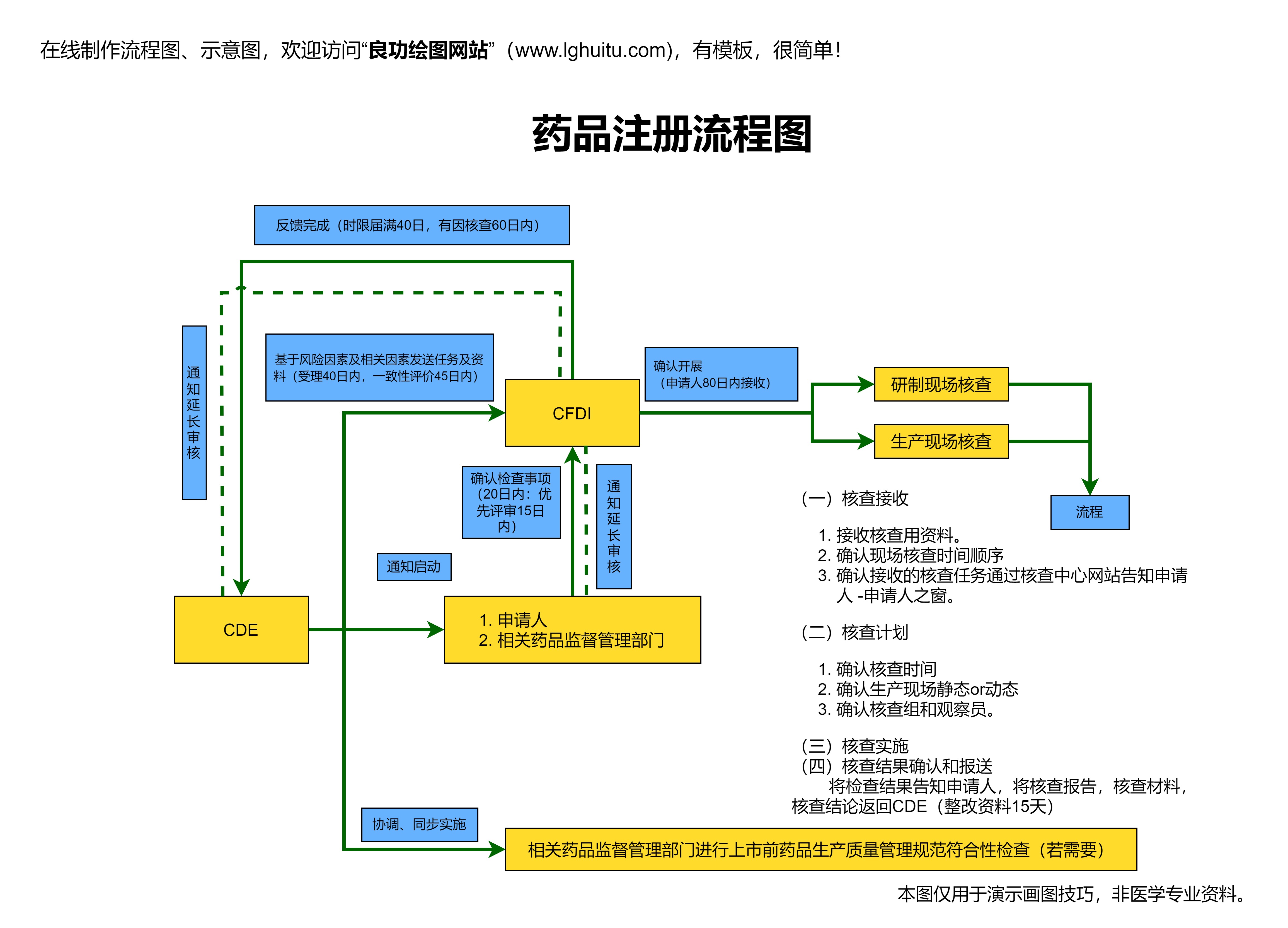
一旦前期资料准备齐全,企业可以向国家药品监督管理局提交注册申请。注册申请包括提交药品的相关资料,并填写申请表格。需要注意的是,药品的注册申请必须符合NMPA的标准,并且要按时提交所有所需的文档,避免因资料不全导致审批延迟。

NMPA收到药品注册申请后,会进行详细审核。审核的内容包括药品的生产工艺、质量控制、临床试验数据、药品标签和说明书等。除了资料审核外,NMPA还可能派遣专家进行现场检查,确保生产厂房符合GMP标准,生产过程符合中国的法规要求。这个阶段是整个申请流程中的核心环节。
在完成资料审核和现场检查后,NMPA会组织相关领域的专家进行评审。专家评审主要对药品的安全性、有效性、质量控制等方面进行严格审核。如果药品通过专家评审,且符合相关规定,NMPA将发放“国药准字号”,并允许药品上市。
即使药品获得了国药准字号,也并不意味着一切完结。药品上市后,企业仍需按规定进行后续的质量监控与报告工作,确保药品在市场上的质量符合标准。如果出现质量问题或不良反应,企业可能会受到监管部门的处罚,甚至面临召回产品的风险。
申请时间长:国药准字号的申请流程涉及多个环节,通常需要一年以上的时间,甚至可能更长。这就要求企业在申请前做好充分的准备,提前规划时间。
申请费用较高:药品注册过程中涉及的费用包括临床试验费用、资料准备费用、专家评审费用等。对许多中小型企业来说,这笔费用可能是一个不小的负担。
市场准入复杂:随着中国药品市场的竞争激烈,获得国药准字号并不代表就能轻松占领市场。企业还需在产品营销、品牌建设、渠道拓展等方面做足功夫。
在了解了国药准字号的基本概念、申请条件及流程后,企业可以更有针对性地开展工作。我们将深入探讨如何优化申请流程,提升申请成功率。