在现代医学和制药技术飞速发展的今天,新药的研发已成为推动全球医疗进步的关键之一。在一个新药从实验室到患者手中的过程中,最为复杂和关键的环节之一便是新药的上市审批。无论是制药公司,还是相关的医药专业人员,理解新药上市审批的全过程,不仅有助于提升产品的市场竞争力,还能确保药品的安全性与有效性符合国家标准。
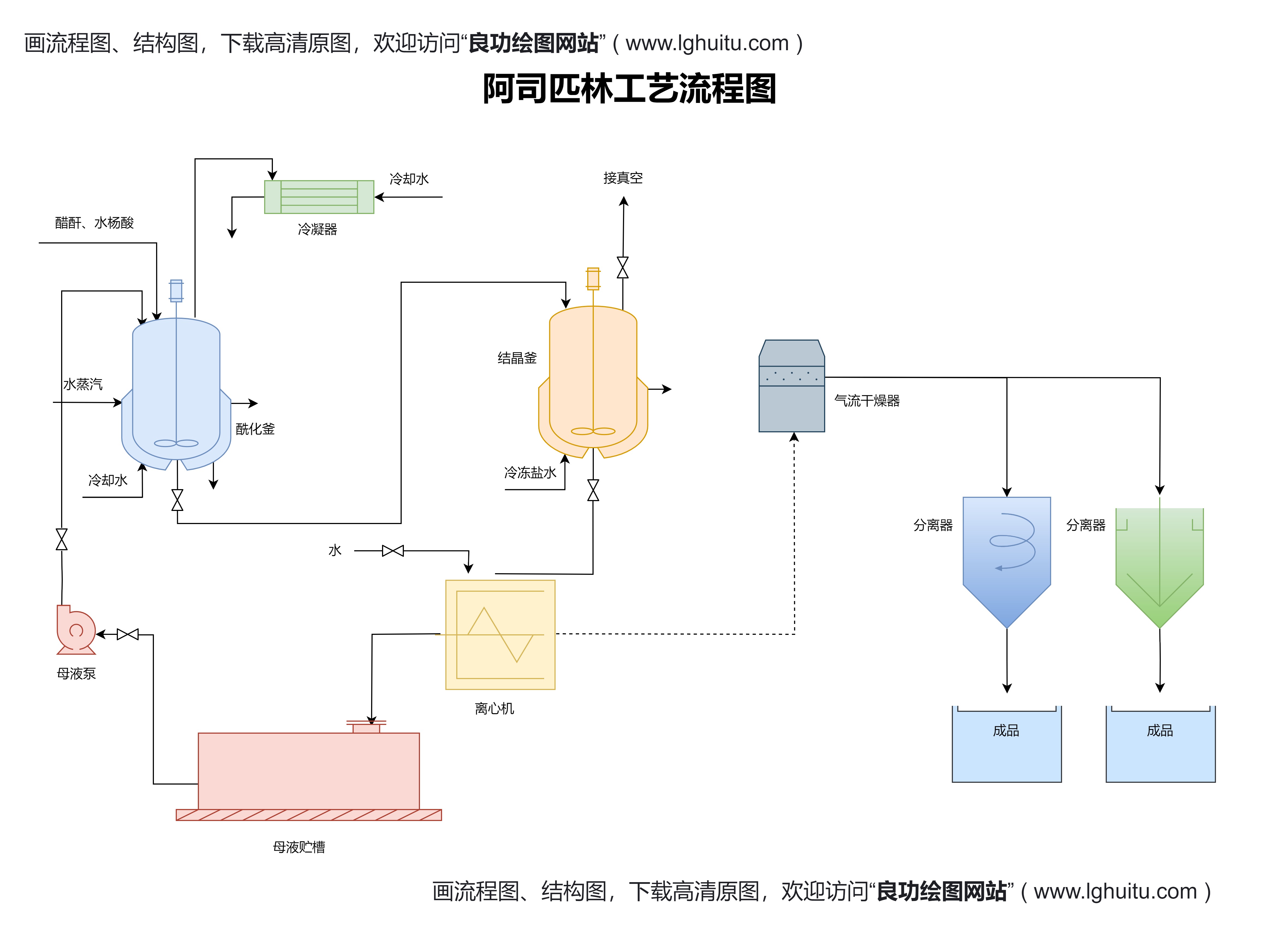
新药的上市审批是一个复杂的多阶段过程,通常涉及临床试验、药品注册、审评审查等多个环节。总体而言,审批流程主要分为以下几个关键步骤:
临床前研究是新药研发的第一步,通常包括动物实验、毒理学研究、药理学研究等。这一阶段的目的是验证药物的安全性和初步的药效,为进入临床试验提供必要的科学数据支持。只有通过临床前研究,才能为临床试验提供充分的理论依据。
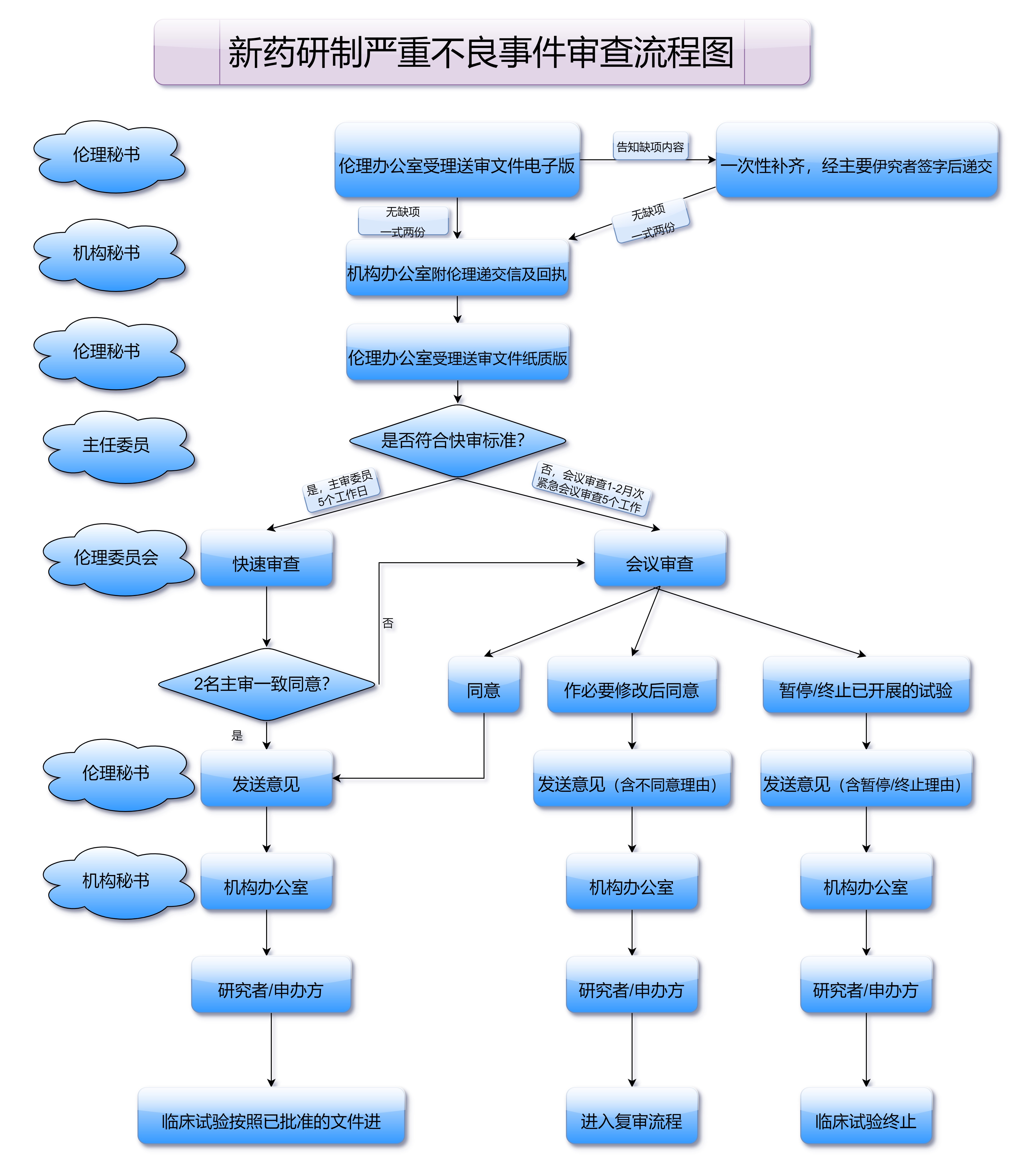
临床前研究完成后,药品研发公司需要向国家药品监管部门提交《临床试验申请》,并在获得批准后,开始开展人类试验。临床试验分为三期,每一阶段都需要通过严格的审核和评估:
第一期临床试验:主要评估药物的安全性、耐受性以及药代动力学。
第二期临床试验:评估药物的初步疗效及剂量的合理性。
第三期临床试验:这是最为关键的阶段,旨在全面评估药物的疗效与安全性,为上市申请提供数据支撑。
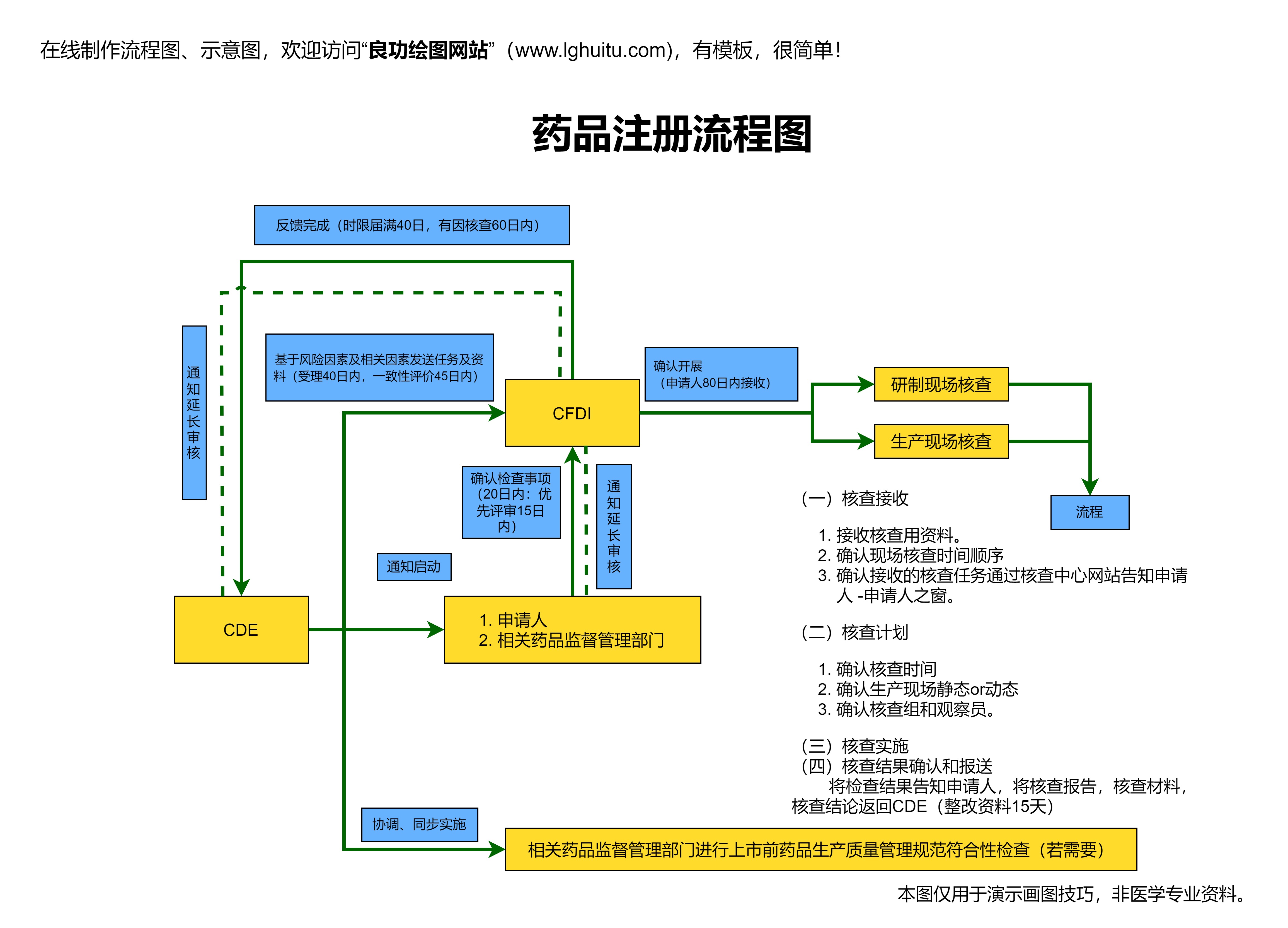
临床试验完成后,制药公司根据三期临床试验的结果,提交《新药上市申请》。药品上市申请需要提交的材料包括临床试验数据、药品生产工艺、质量控制标准等,监管部门将根据这些材料对药品的安全性、有效性及质量进行综合评审。

药品上市申请提交后,监管部门会进行详细的审评审查。审评的内容包括药品的质量、临床试验数据、药物的生产工艺、市场需求等因素。这个阶段的时间长度因药品种类、复杂程度等因素而有所不同。
如果审评通过,药品会获得药品注册证书,并正式进入市场。药品的上市意味着它可以在特定的地区或国家进行销售和使用。药品上市后仍然需要进行长期的跟踪研究,监测药品在广泛使用中的安全性和效果。
新药上市审批过程中有多个关键环节需要特别关注,其中包括药品注册申请、临床试验数据的提交与审查、药品质量控制标准的设置等。每一个环节都可能对药品的上市进程产生重大影响。
药品注册申请是药品上市的起点,申请材料的规范性直接影响审批速度。为了提高审批效率,制药公司必须确保提交的材料准确、完整且符合法规要求。材料的遗漏或不规范可能导致审批流程延误,甚至被退回重新提交。
临床试验是审批过程中的重中之重。药品的疗效和安全性需要通过严格的临床试验来验证。每一阶段的试验结果都需要经过专家审查,且必须符合国家和国际药品监管标准。试验数据的可靠性是药品能否顺利上市的关键。
药品的生产工艺和质量控制标准直接关系到药品的品质与市场竞争力。监管部门会对药品的生产过程进行严格审查,确保药品在生产过程中的每一步都符合GMP(良好生产规范)要求,保障药品的质量和一致性。
审批过程中,药品生产商还需要确保药品能够满足相关国家的标准和法律要求,进行详尽的市场调研和成本效益分析。除此之外,药品在上市后依然需要进行风险管理和长期监控。以下是新药上市后的后续工作及市场准入的进一步要求。
即使新药成功上市,监管部门对药品的关注并未结束。药品上市后,制药公司必须进行药品的后续监控,包括不良反应的监测、药品的临床使用反馈以及生产过程的持续审查。这一过程被称为“上市后监控”或“药物警戒”。
药品上市后,制药公司需要定期向监管部门报告药品的使用情况,尤其是可能出现的不良反应。通过不良反应的监测,能够及早发现药品的潜在风险,并采取相应的措施,如调整药品使用说明书,甚至是暂停或撤销药品的上市。
药品上市后,临床使用者的反馈至关重要。制药公司应当通过渠道收集医生和患者的使用体验,确保药品能够在不同患者群体中发挥最佳疗效。收集的反馈信息还能够为未来的药品改进和新的适应症开发提供参考。
药品的生产过程中,药品生产企业需要保持GMP标准,并定期接受监管部门的检查。生产工艺、设备设施、人员管理等各方面必须符合标准,确保药品的质量始终保持稳定。
随着技术的进步和医学领域的不断发展,新药的审批流程也在不断创新。数字化和人工智能的引入,使得药品的研发与审批流程更加高效和精确。全球范围内,多个国家和地区正在探索更加灵活和在线画图工具的审批路径,以应对新药研发速度加快和市场需求日益增长的挑战。
新药上市审批的过程不仅关系到药品的安全性和有效性,还直接影响患者的治疗选择。各国药监部门在确保药品安全的也在努力优化审批流程,推动创新药物早日进入市场,为全球患者带来更多福音。