新药的研发过程是一个漫长而复杂的旅程,从药物的初步发现到临床研究,再到最终的上市,每一步都关乎人类健康的福祉。尤其是在新药的临床研究审批过程中,严谨的程序和高标准的要求确保了药物的安全性和有效性,而这一过程也正是药物能够顺利进入市场的重要保障。
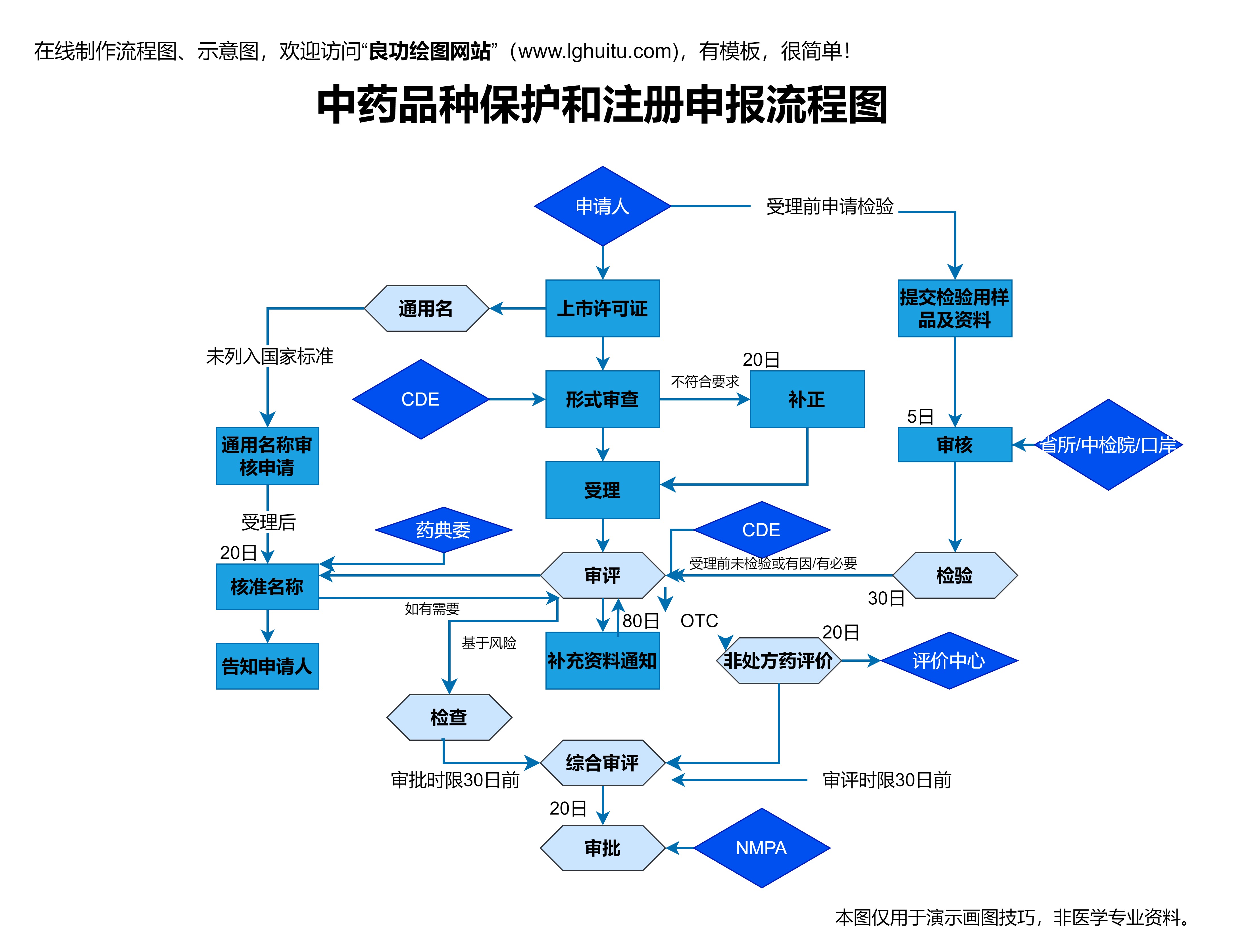
临床研究审批流程是新药研发中的核心环节,它不仅需要经过多层次的审核与批准,更是科学与伦理相结合的产物。通常来说,新药的临床研究审批流程分为多个阶段,包括临床试验申请(IND申请)、临床研究的实施、数据分析与审查、以及最后的上市审批等。
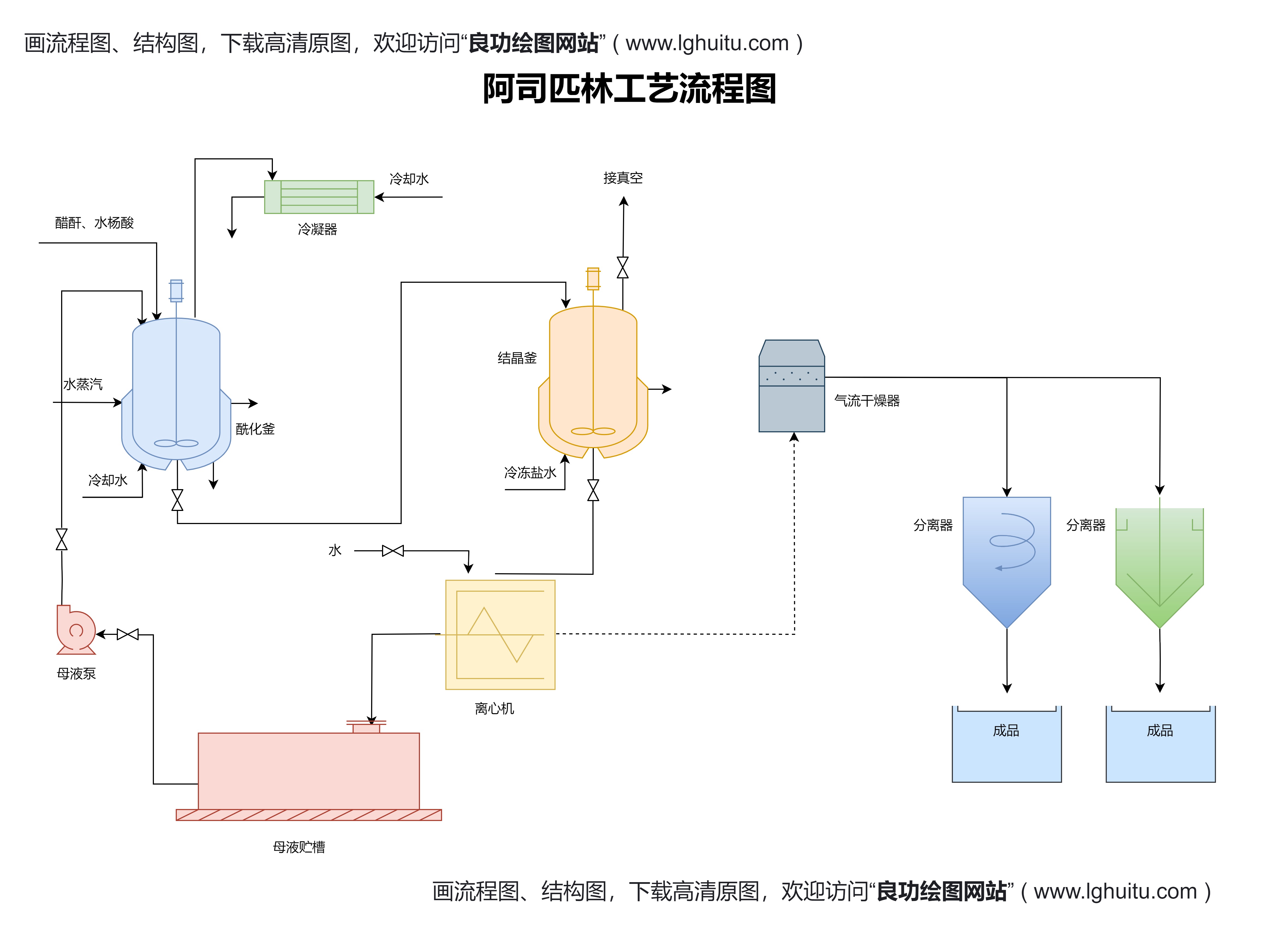
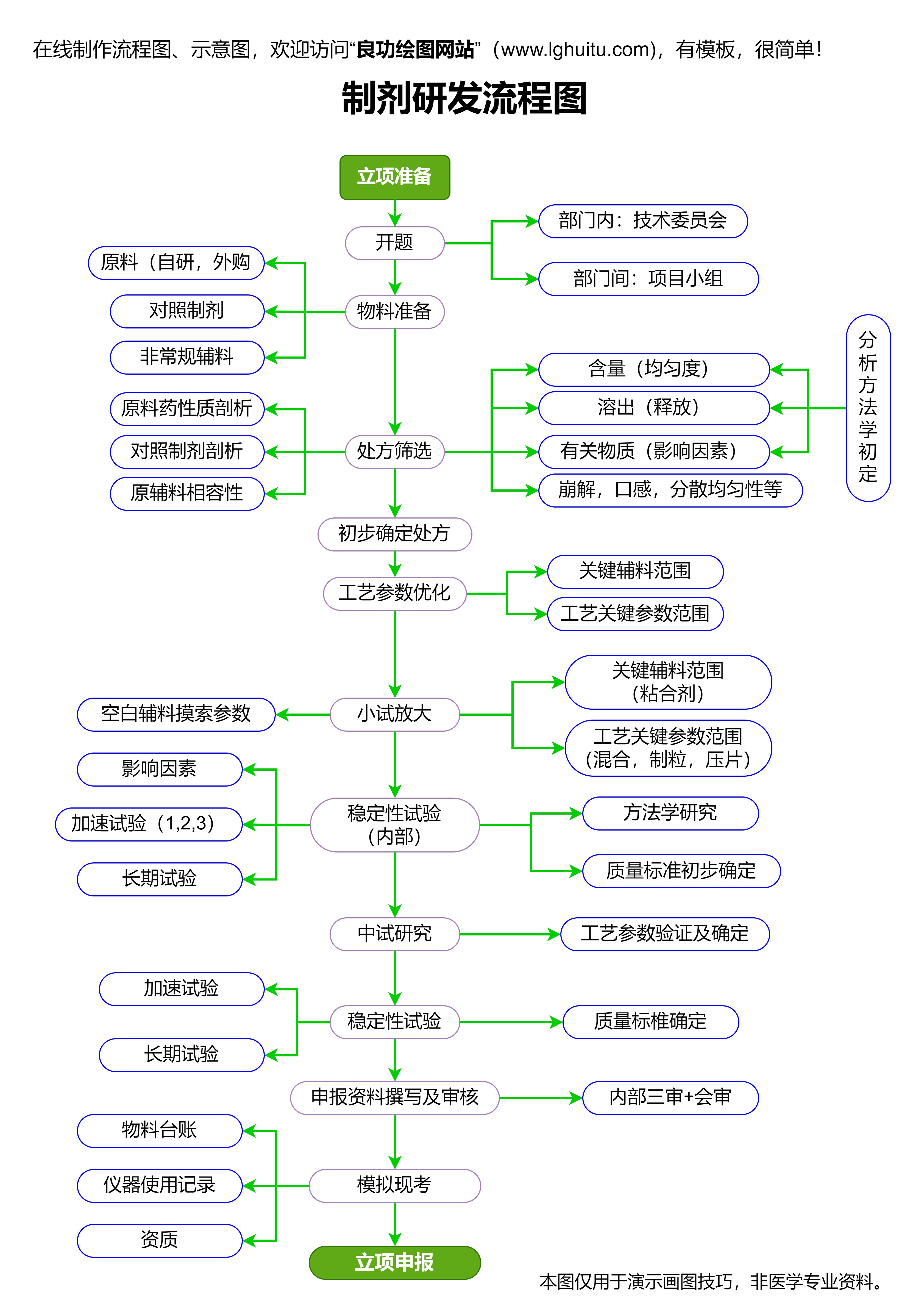
新药在临床研究之前,需要经过一系列的前期研究。通常,药品研发公司会进行体外实验、动物实验等,以确认药物的基本安全性和药理作用。这一阶段的成果为后续的临床试验奠定基础。
在完成这些初步研究后,制药公司就可以向药品监管机构提交临床试验申请(IND申请)。这一申请是新药临床试验的正式启动前提,申请内容包括药物的非临床研究数据、临床试验的方案、以及预期的药物治疗效果等。只有当监管机构批准了该申请,制药公司才能进行人类临床试验。
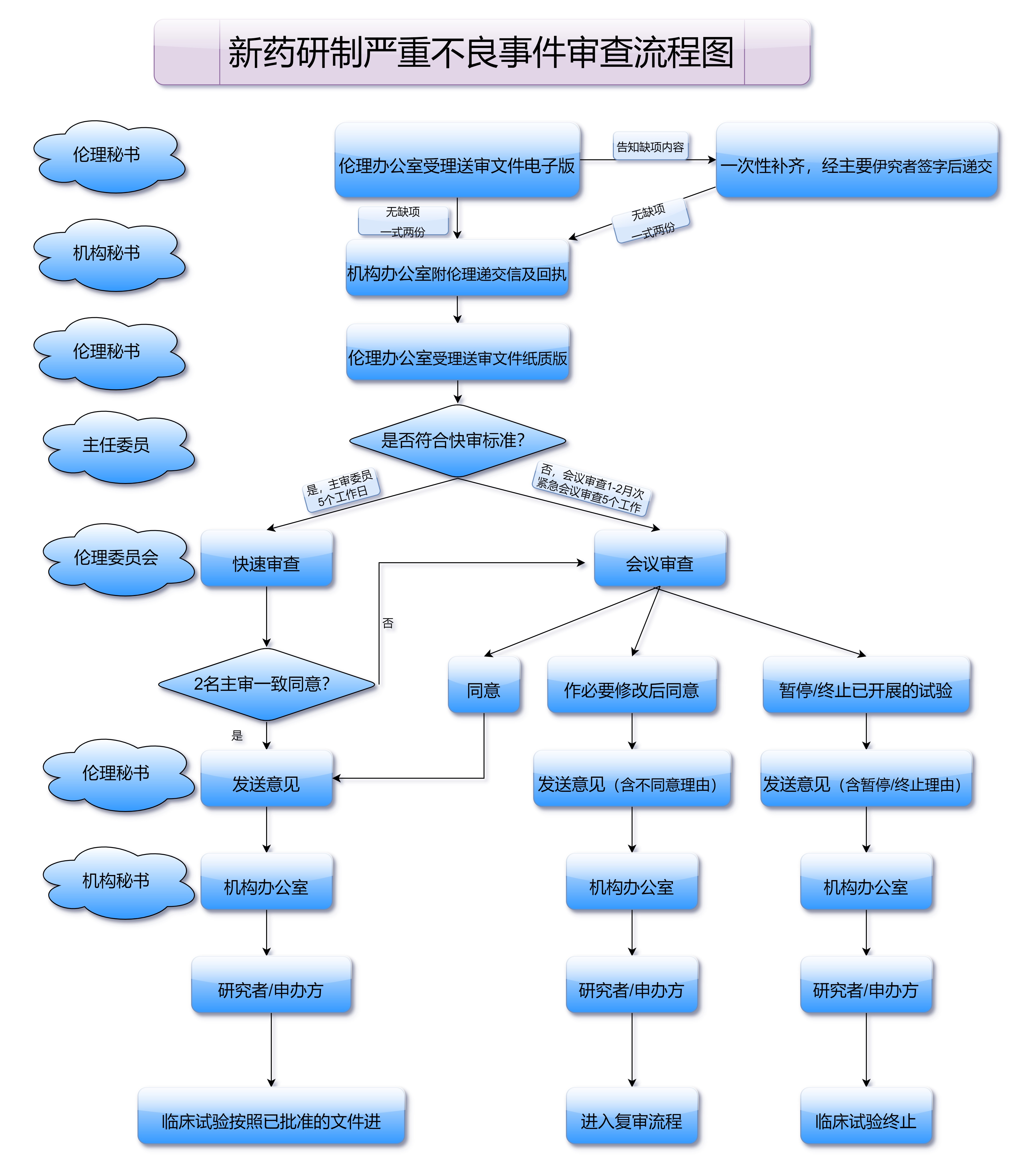
临床试验一旦启动,就进入了严格的监控和数据收集阶段。在这个过程中,药品监管部门会持续对试验进程进行跟踪,确保研究过程符合法律法规要求,并且保证受试者的安全。在试验过程中,临床数据的采集与分析尤为重要,任何不合规的行为或数据都可能导致临床试验被暂停或终止。因此,临床研究的执行不仅是对科学的挑战,更是对责任的承诺。

随着临床试验的深入,数据逐渐积累,研究人员会对药物的有效性和安全性进行详细分析。这一过程是新药是否能顺利上市的关键。制药公司需要根据试验结果,准备详细的报告,并提交给监管机构进行审查。在这一阶段,药品监管机构将对临床试验数据进行严谨的评估,决定药物是否可以进入下一阶段——上市申请。
新药的临床研究审批过程中,上市申请(NDA申请)是至关重要的环节。上市申请的材料不仅要包括药物的临床试验数据,还需要包括药品生产工艺、质量控制标准、药品的包装与标签、药物的疗效与副作用等多方面的信息。监管机构会在收到这些材料后,进行全面审查,确保药物符合所有安全、有效和质量标准。
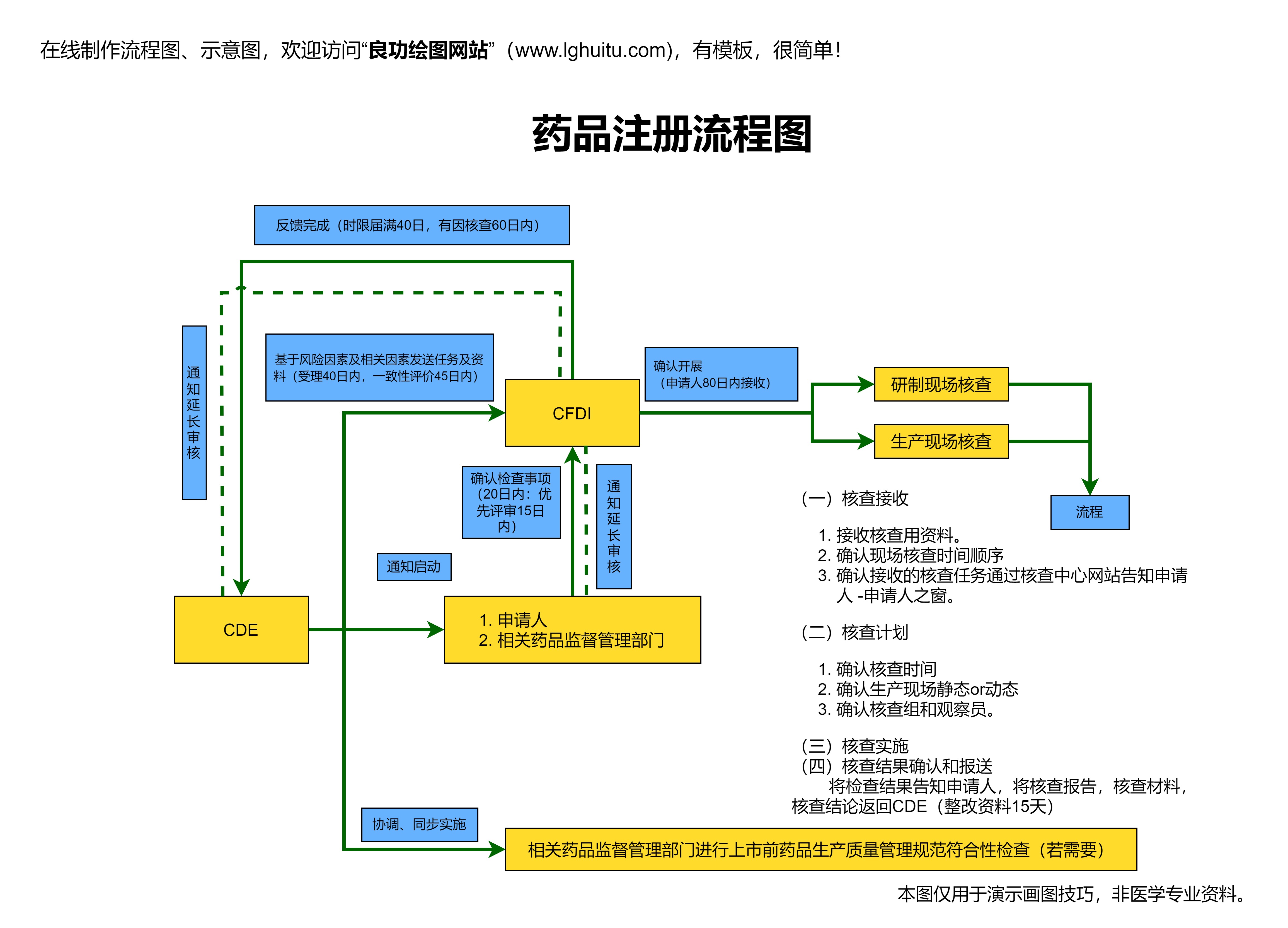
通常,监管机构会在审查过程中邀请专家组进行会议评审,专家们会根据临床试验数据、药品质量控制标准等进行详细讨论,确保药品的上市不会对公众健康造成危害。在这一阶段,药品的临床研究审批不仅仅是一个技术性的审查过程,更涉及到伦理和社会责任的考量。药物的最终批准不仅是科学成果的体现,更是公众信任和安全的保障。
若药品顺利通过审批并获得上市许可,那么它便可以进入市场,开始为患者提供治疗。这时,药品的生产、销售和使用将受到监管机构的持续监控。药品上市后的不良反应监测(ADR监测)也是新药审批流程中的重要一环。即使药品已经上市,监管机构仍会继续对其进行监控,确保药品在市场上使用过程中的安全性。若发现新的不良反应,药品的使用可能会受到限制,甚至撤市。
新药的临床研究审批流程不仅仅是一个繁琐的程序,更是保证药品安全性、有效性与质量的根本保障。对于制药公司来说,这一流程是他们通向市场和患者的桥梁,也是药物创新的一个重要标志。对于公众而言,严格的审批流程为他们提供了更安全、更可靠的药品选择。而随着技术的发展与法规的不断完善,未来的新药研发与审批将更加高效、透明,能够更快地满足患者的治疗需求。
新药临床研究的审批流程是一项严谨且复杂的任务,涉及的每一个环节都至关重要。从临床试验的设计到数据分析,再到最终的上市审批,每一步都需要遵循科学与伦理的双重标准。这不仅仅是药品从实验室到市场的转变,更是对患者健康的责任与承诺。