新药研发,是制药行业最为复杂且充满挑战的过程之一。无论是药物的初步发现、临床试验,还是新药的上市,背后都凝聚了无数科研人员的努力与智慧。在这一过程中,新药申请的流程则是至关重要的一环,它直接决定了药品能否进入市场,为患者带来治疗的希望。今天,我们就来详细探讨一下新药申请的流程,带您了解每一个关键环节,帮助制药企业顺利应对各种挑战。
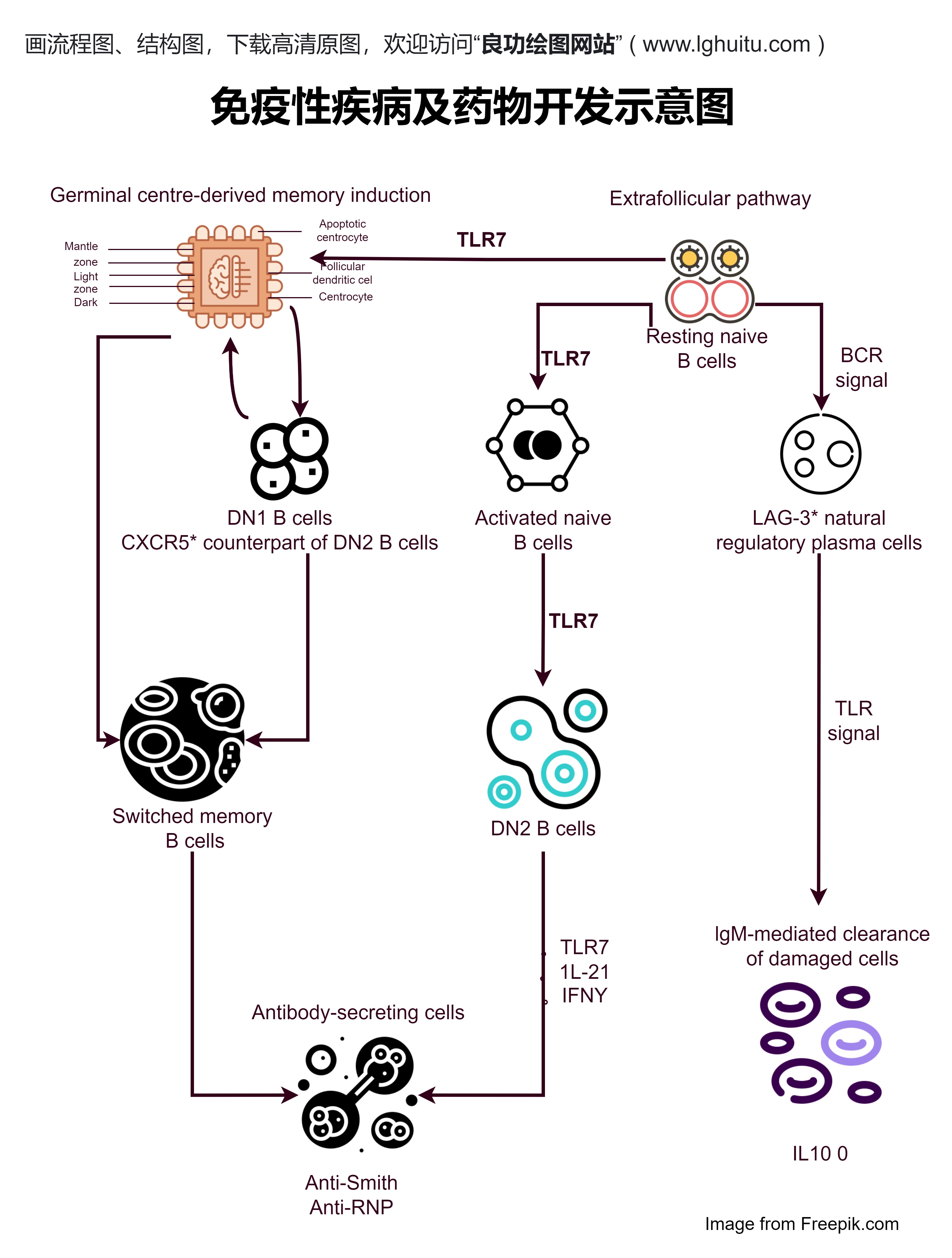
新药申请的流程大致分为四个阶段:临床前研究、临床试验、审批申请、以及上市后的监管。在这些环节中,每一步都需要企业与监管部门密切合作,确保药物的安全性、有效性与质量。
新药申请的第一步通常是临床前研究,也就是在药物进入人体试验之前,科学家通过一系列的实验室研究,评估药物的安全性与有效性。在这一阶段,研发团队将首先对候选药物进行化学、药理毒理以及药代动力学等研究,以确保药物不会对动物或人体产生致命毒性。
在临床前研究阶段,还需要大量的动物实验数据来验证药物的初步效果。这些研究数据将为后续的临床试验奠定基础,并为药品注册提供可靠的科学依据。对于大部分新药,临床前研究的完成通常需要2到3年的时间。
临床试验是新药研发过程中最为关键的环节之一,也是药物上市前的最后一道“安全门”。临床试验通常分为三期:I期、II期和III期,每一期都有不同的研究目标与要求。
I期临床试验,通常是在少量健康志愿者中进行,主要目的是评估药物的安全性与耐受性。该阶段的研究非常严格,药物的剂量、用药方式、潜在副作用等都会得到详细监测。
II期临床试验则通常在小范围的患者群体中开展,重点在于评估药物的疗效和安全性。这一阶段的结果将直接影响药物是否进入III期临床试验。
III期临床试验则是最为全面的评估阶段,通常是在多中心、大规模患者群体中进行,主要目的是进一步确认药物的疗效与安全性,并为药品上市提供最具权威的数据支持。III期试验的成功与否将直接决定药物是否能够进入市场。
每个临床试验的阶段都需要获得监管机构的批准,特别是在中国,药品的临床试验必须提交给国家药品监督管理局(NMPA)进行审查,获得临床试验批件。这个审批过程对新药能否顺利进入临床试验至关重要。
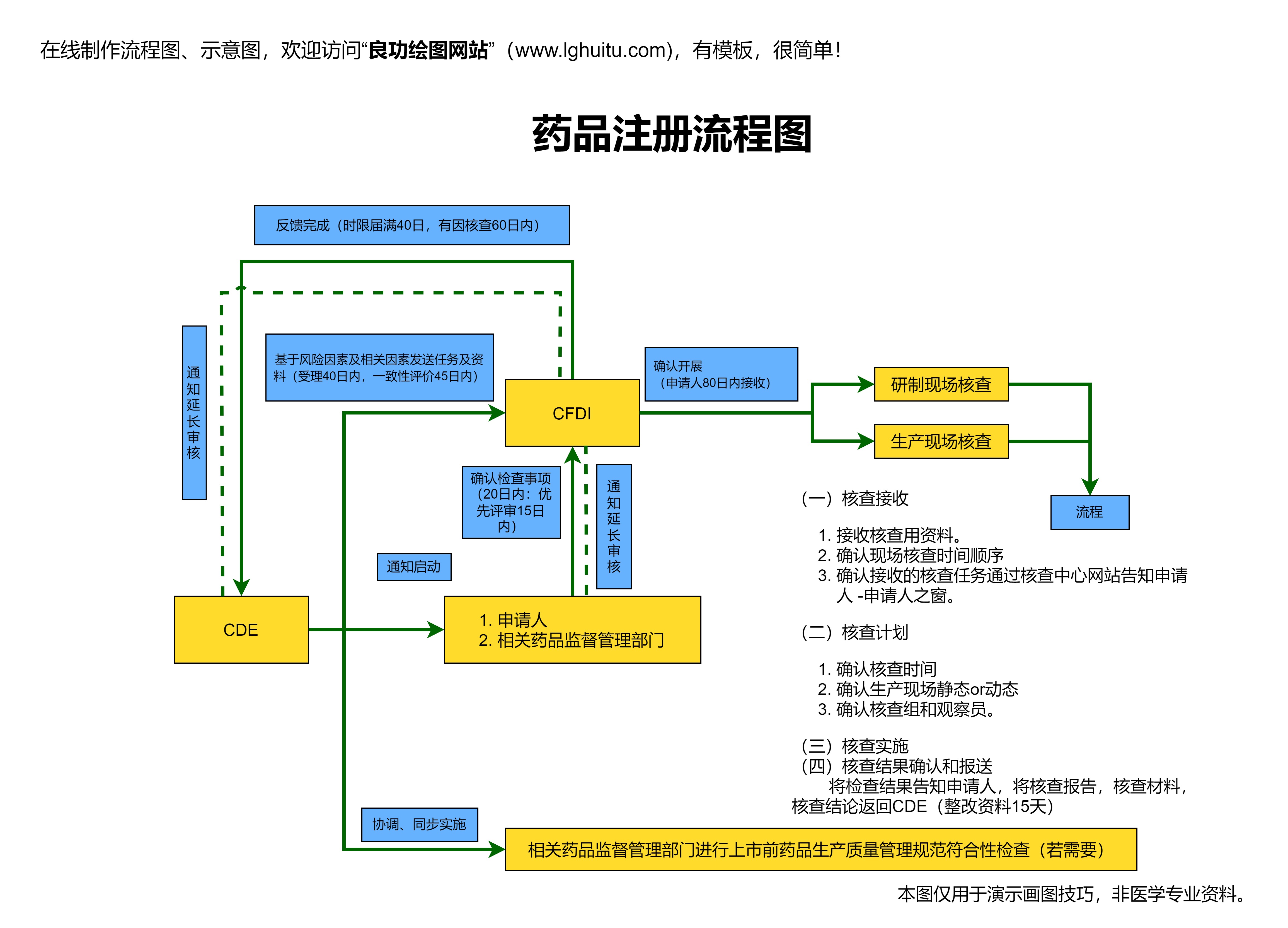
当临床试验取得了令人满意的结果后,制药公司便可以着手准备药品上市前的审批申请。这时,制药公司需要将新药的研究数据、临床试验报告、药物生产流程等详细资料提交给监管机构审查。不同国家和地区的审批程序有所不同,但大致流程相似。
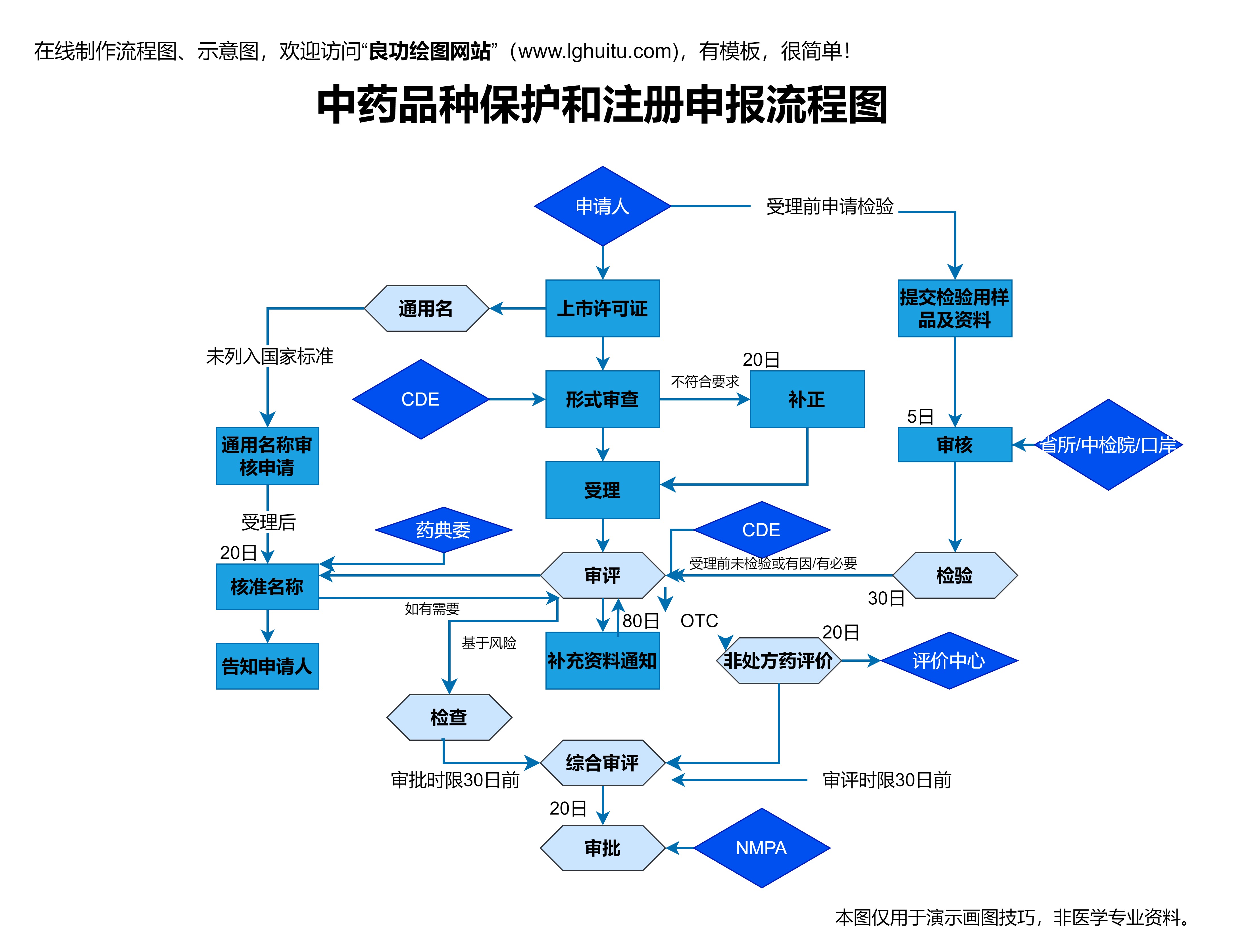
在中国,药品的审批申请需要向国家药品监督管理局(NMPA)提交药品注册申请。NMPA将对所有提交的资料进行严格的审查,包括药品的安全性、有效性、质量控制标准以及生产过程等。该过程通常需要一段时间,审批周期视药品的复杂程度与研究数据的完备性而定。
NMPA还会要求提交有关药品的上市后风险管理计划,并确保制药企业在药品上市后的长期跟踪与监测。只有所有的审批流程都通过,药品才能顺利获得上市许可,进入市场。
新药一旦获得上市许可,并进入市场销售,监管机构并不会放松对药品的监管。实际上,新药的上市后监管是一个持续不断的过程,目的是确保药品在使用过程中始终保持高效与安全。
上市后监管包括药品的不良反应监测、药品质量的持续检查以及药品使用的风险评估等。制药公司需定期向监管机构报告药品的使用情况、患者的不良反应数据,并根据情况调整药品的说明书、使用剂量以及使用禁忌等。
在全球范围内,许多国家和地区都有严格的药品上市后监测制度。例如,在美国,FDA要求制药公司每年提交一次药品的不良反应报告。在中国,药品的不良反应报告制度也日益完善,并由国家药品监督管理局(NMPA)进行严格监管。
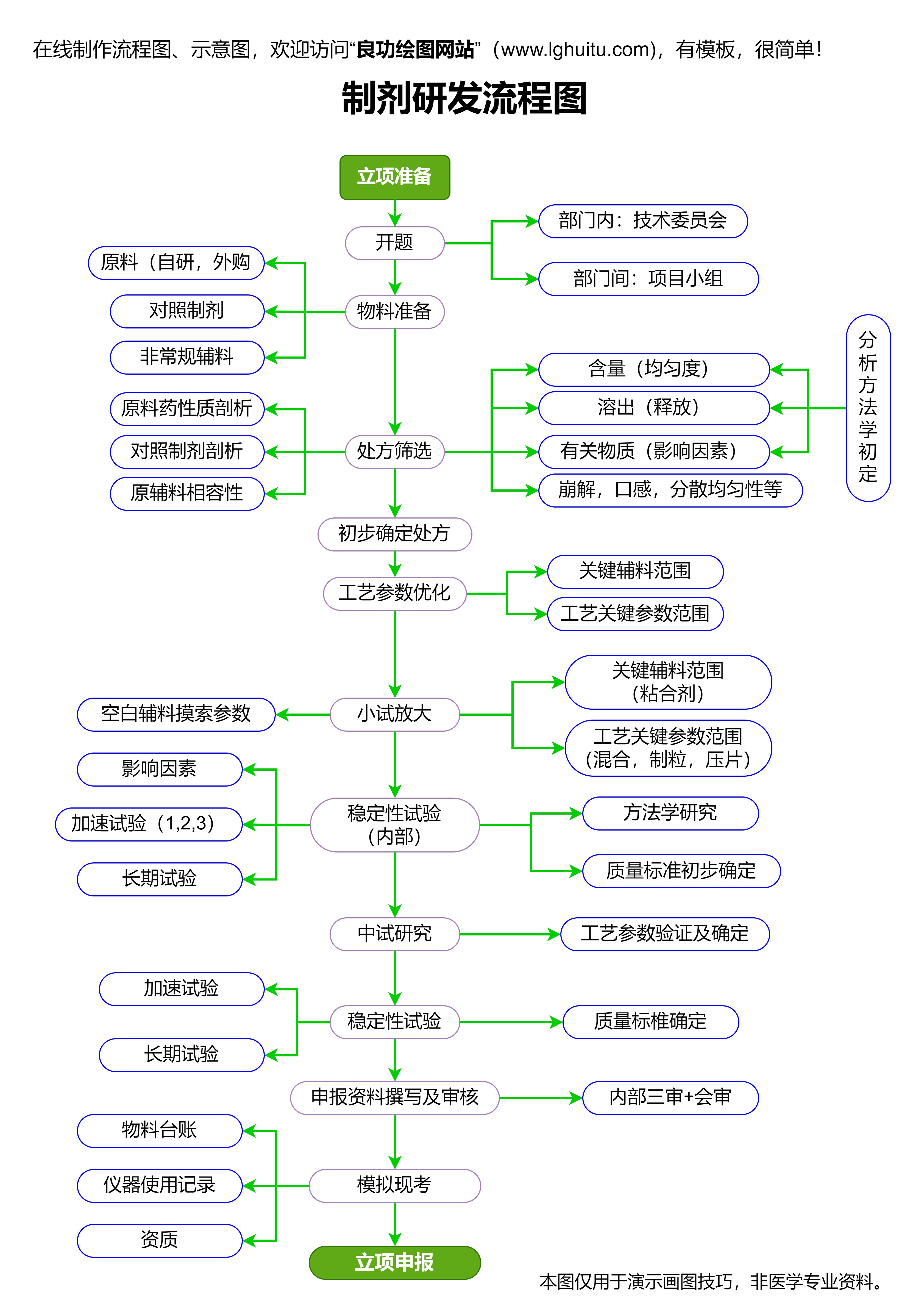
新药申请流程的每一个环节都充满挑战。对于制药公司而言,如何高效地完成临床前研究、临床试验、审批申请,以及如何在上市后进行有效的风险监控,都是需要攻克的难题。而且,药品的研发不仅是科学问题,还涉及到巨大的资金投入与市场风险,因此,企业在这条路上的每一步都需要慎之又慎。

尽管如此,新药的申请与上市依然充满机遇。随着全球医疗需求的不断增加,越来越多的制药公司和科研团队涌入到新药研发的战场。尤其是在生物制药、基因治疗、免疫治疗等前沿领域,新药的研发前景广阔,成功通过申请的机会也越来越多。
新药申请的流程是一个复杂且充满挑战的过程,但也是药物创新与医疗发展的关键所在。从临床前研究到临床试验,再到审批申请与上市后的监管,每一环节都至关重要。企业只有通过精细的科学研究与严格的流程管理,才能最终打破市场壁垒,推出满足患者需求的创新药物。对于制药企业来说,掌握新药申请的每一个环节,不仅是成功的必经之路,更是赢得市场竞争优势的重要法宝。