在中国,药品进入市场前必须经过严格的审批流程,确保药品的质量、安全性和有效性。为了获得合法销售资格,药品需要通过国家食品药品监督管理局(NMPA)的审批,并获得“国药准字号”。这个准字号是药品合法上市的关键标志,也是消费者和医生判断药品安全性的重要依据。申请国药准字号到底需要满足哪些条件呢?本文将为您详细解析。
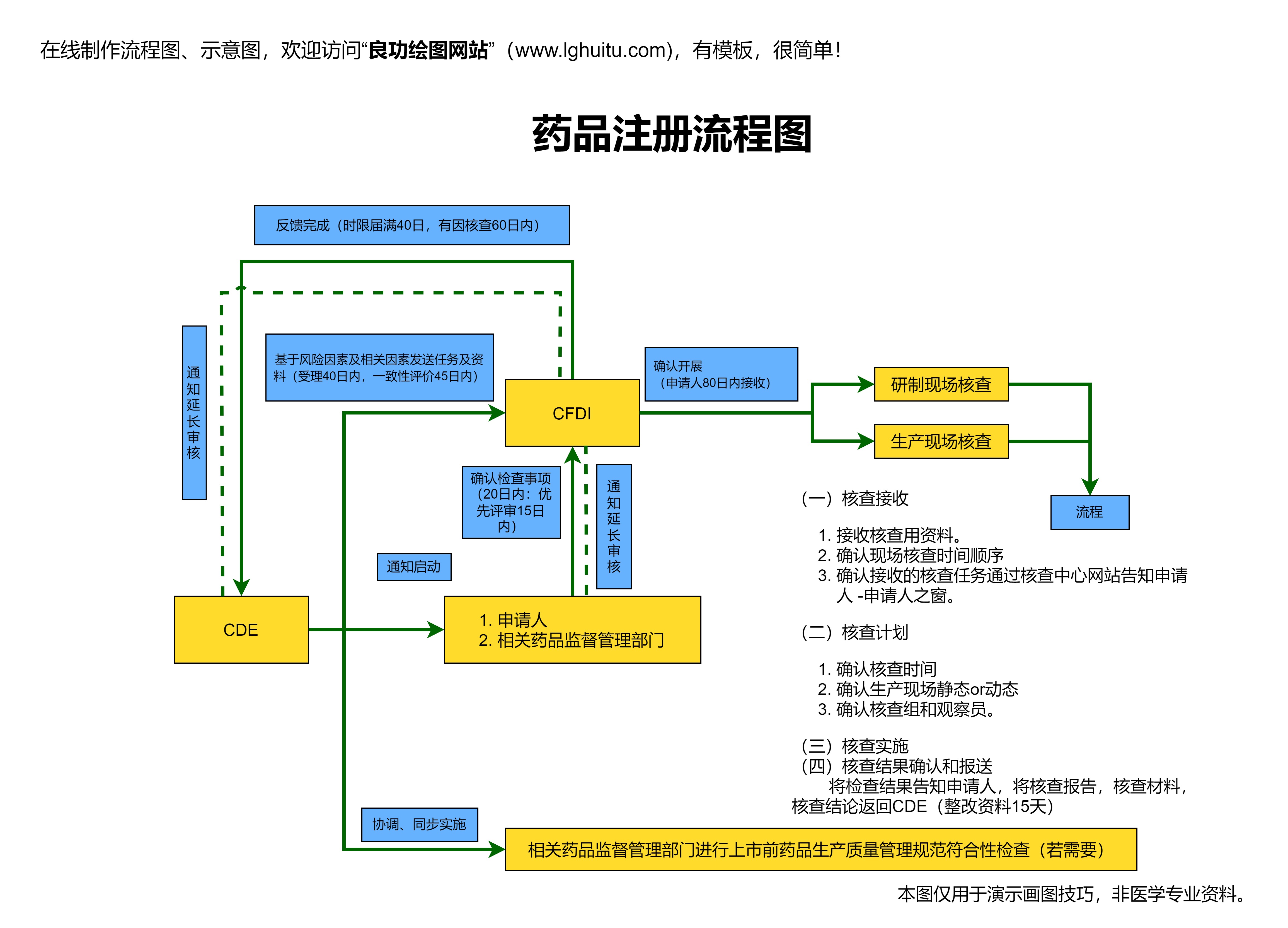
申请国药准字号的前提条件是药品必须符合《药品管理法》及相关法规的要求,确保药品符合药品生产和质量管理的基本标准。药品在申请过程中需要提供详细的科研数据,包括临床试验数据、药理学与毒理学研究结果等。这些数据是证明药品安全性和有效性的依据,也是国家监管部门审批的重要依据。
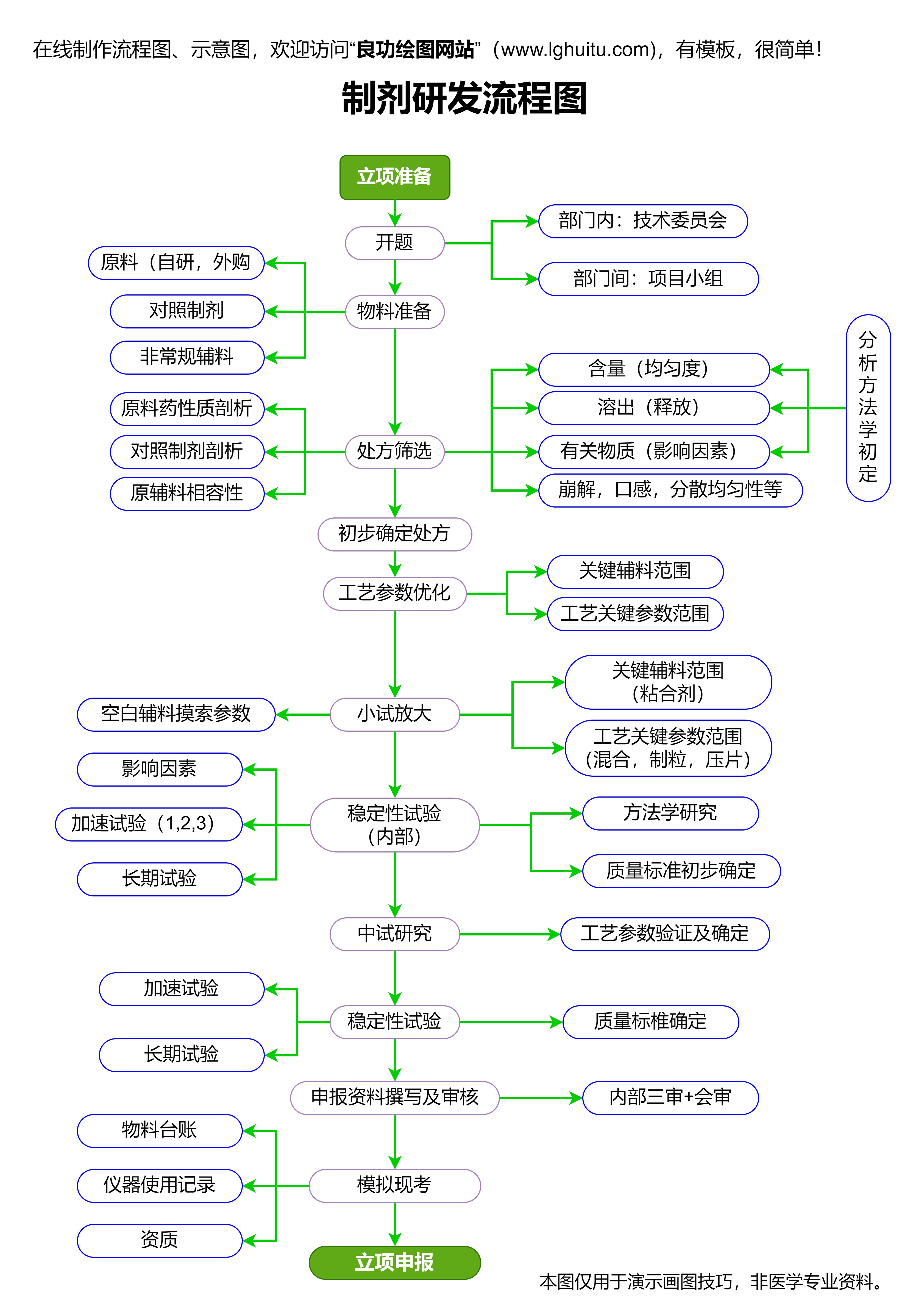
药品的生产企业必须具备符合国家要求的生产条件。药品生产企业需要拥有符合GMP(药品生产质量管理规范)标准的生产设施和设备,且生产过程必须符合国家食品药品监督管理局的监管要求。药品的生产环节必须严格按照规范进行,以确保每一批次药品的质量都能稳定且可控。
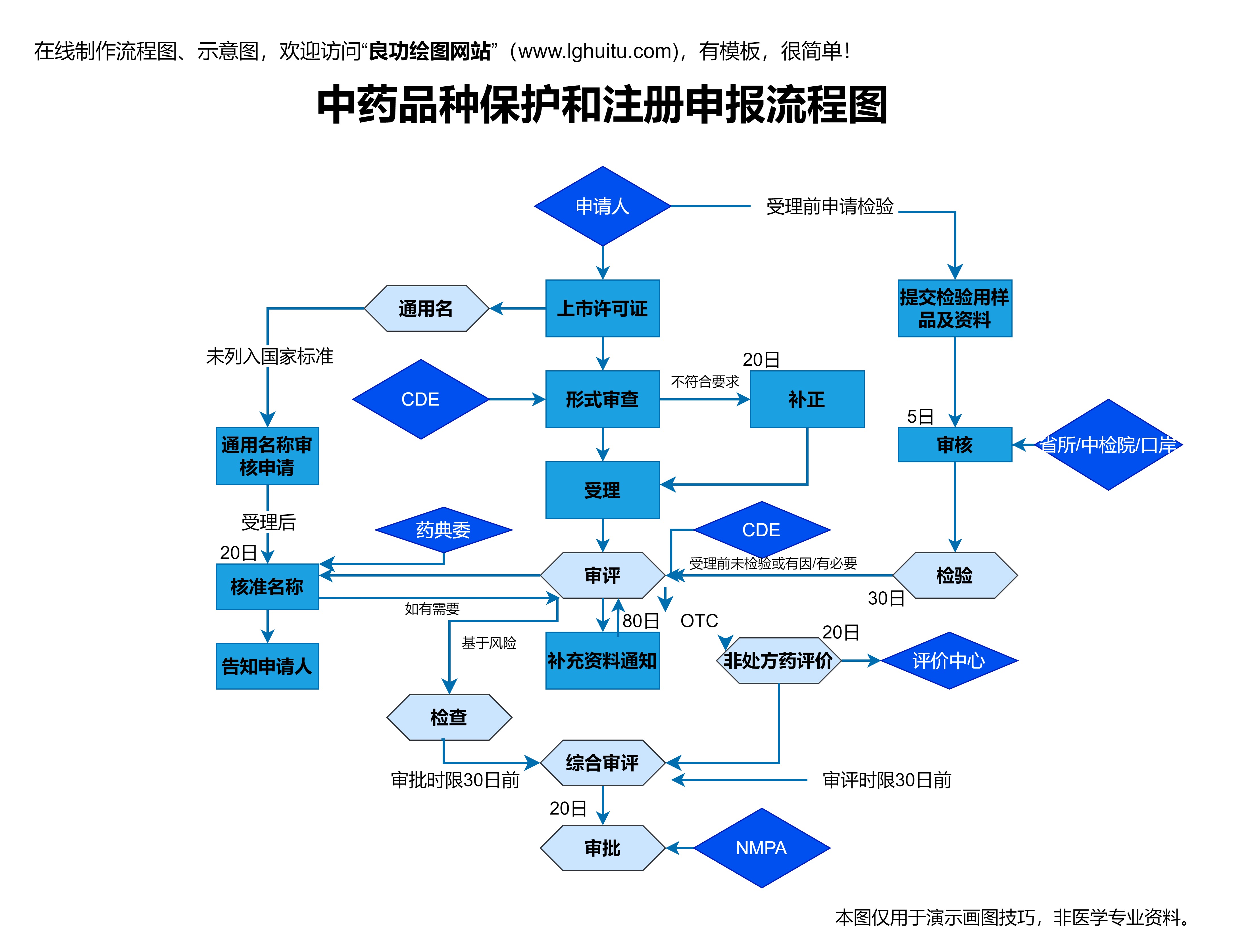
药品的注册申请必须由药品生产企业在规定的时间内提交完整的资料。申请资料包括药品的基本信息、生产工艺、质量标准、临床试验报告、药品标签等。这些资料的提交必须符合国家药品注册管理的相关规定,任何一个环节的疏漏都有可能导致申请失败。
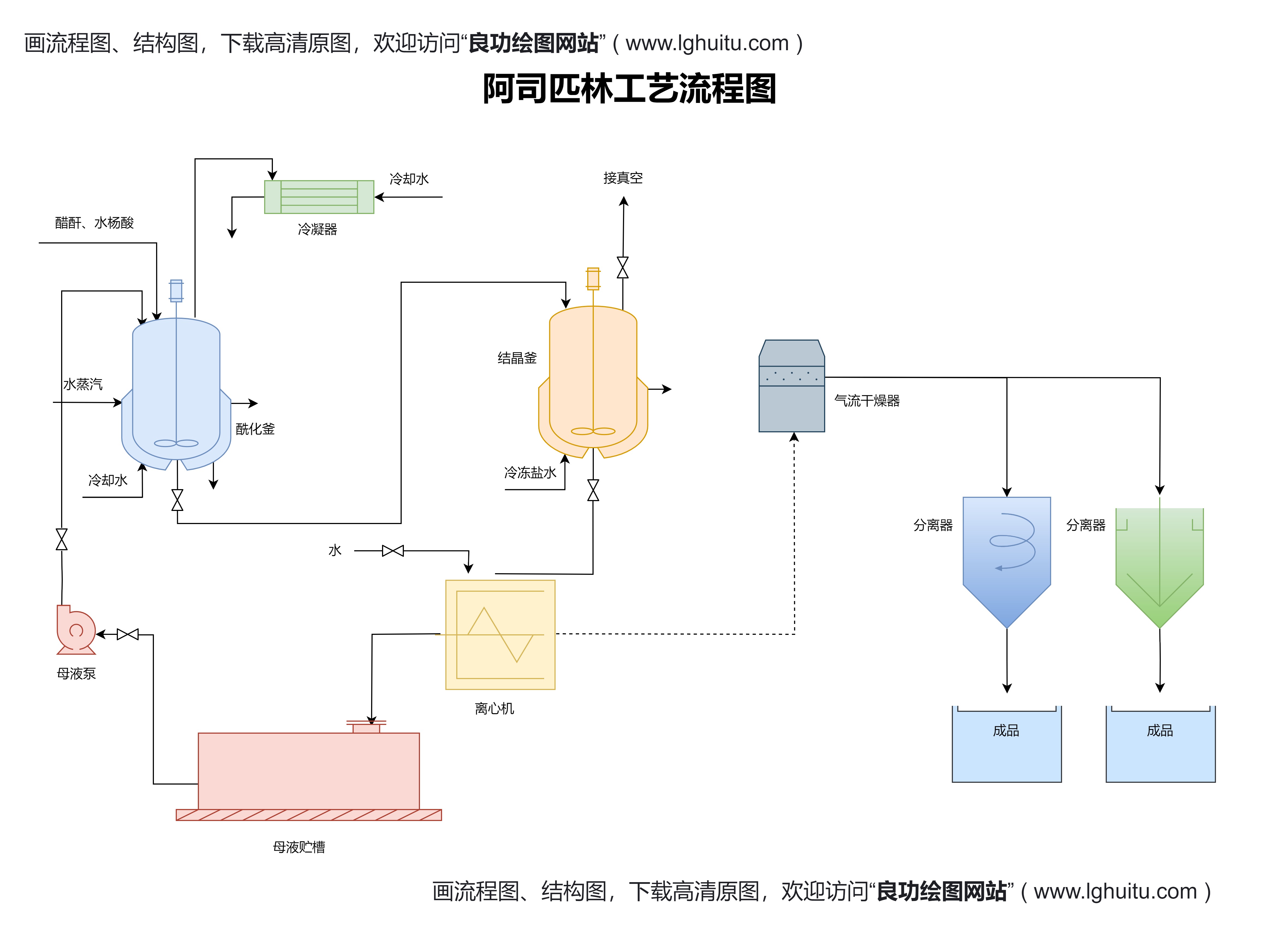
在药品的临床试验阶段,申请国药准字号的药品通常需要通过多个阶段的临床试验,包括I期、II期和III期临床试验。这些临床试验需要在符合伦理要求的条件下进行,且所有的试验数据必须真实有效。尤其是对于新药或改良药品,必须通过严格的临床数据验证其安全性和有效性。
除了科研数据和生产标准,药品的市场需求也是审批过程中重要的考量因素。国家药品监督管理部门会根据药品的临床适应症、市场前景以及患者的实际需求等因素进行综合评估。在某些特殊情况下,国家还会根据药品的特殊性给予加速审批或绿色通道,以便快速满足紧急医疗需求。

需要特别强调的是,药品在获得国药准字号后,并不意味着可以立即进入市场。药品还需要在销售过程中接受持续的监管,确保药品在流通和使用中的质量始终符合国家标准。
我们将详细探讨申请国药准字号的具体流程和注意事项,帮助药企更好地准备相关材料,顺利通过审批。
申请国药准字号的流程通常可以分为几个主要步骤。首先是药品的注册申请准备阶段。在这一阶段,药品生产企业需要向国家食品药品监督管理局提交药品注册申请书及相关资料。申请资料的完整性和准确性是审批成功的关键,因此企业需要在这一阶段做好充分的准备。资料提交后,药品注册申请将进入审查阶段。
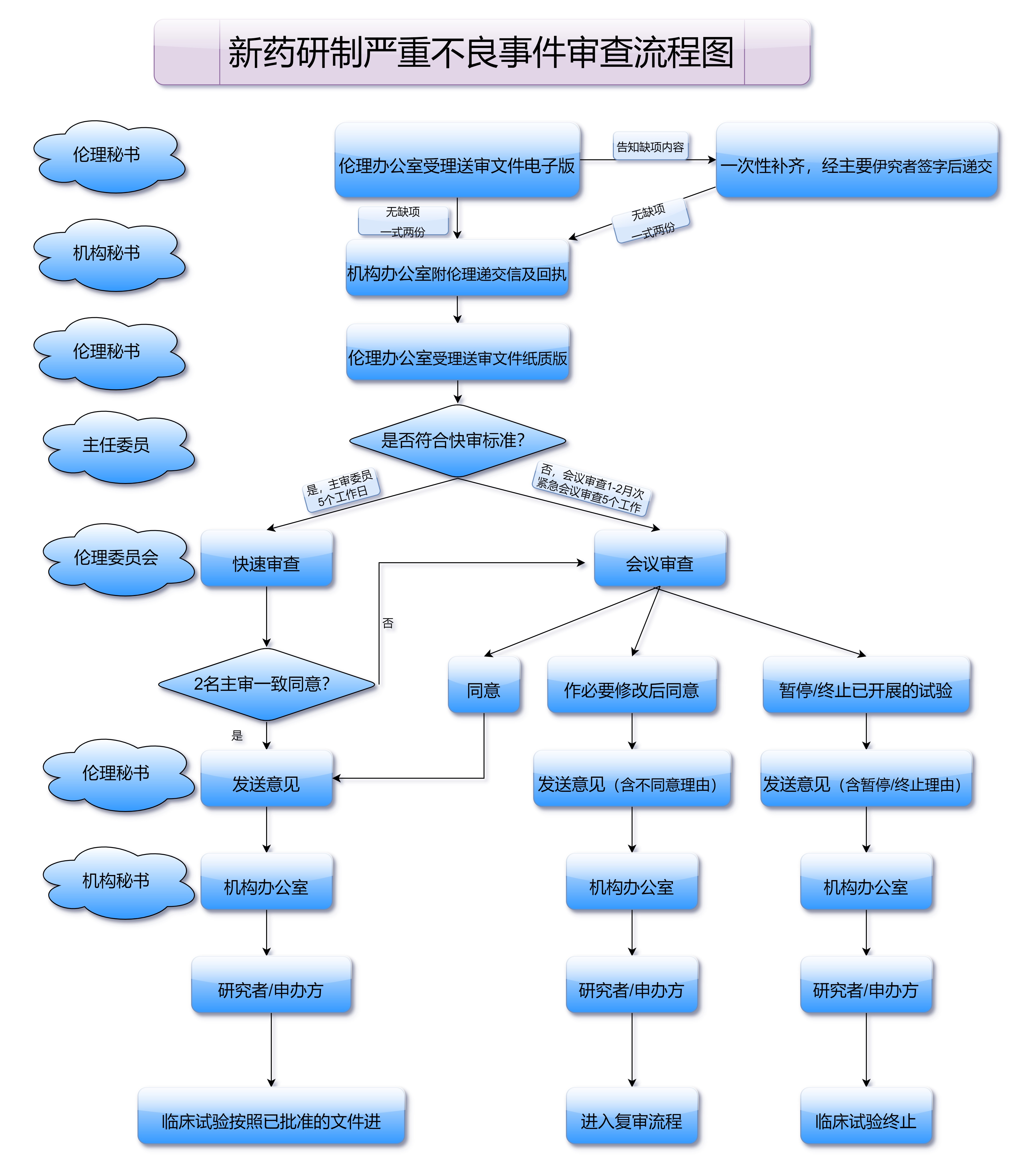
审查阶段分为行政审查和技术审查两个部分。行政审查主要审核提交资料的完整性和合规性,确保所有资料符合国家药品注册的规定。技术审查则是重点审核药品的科学性、临床数据的有效性、生产工艺的合理性等方面。技术审查的核心目的是确保药品的安全性、有效性和质量可控性。
在审查阶段,审查人员会对药品的临床数据、质量标准、生产工艺等进行深入分析和评估,必要时还会要求申请企业提供补充资料或进行现场核查。审查通过后,国家药品监督管理局会发布正式的批准文件,并授予药品“国药准字号”,药品可以合法上市销售。
除了常规的审批流程,药品企业还需要注意以下几点。在药品的临床试验过程中,企业应严格遵循伦理要求,确保临床数据的真实性和完整性。药品的生产设施也必须定期接受药监部门的检查,确保生产过程的稳定性和可控性。
企业还应关注药品上市后的市场监测与管理。即使获得了“国药准字号”,药品在市场上的流通和使用也需要不断地接受药监部门的监督。企业应根据药品的市场表现和临床反馈,及时对药品进行风险评估,并采取必要的措施来保障患者的用药安全。
申请国药准字号的过程是一个复杂且严格的过程,需要药品企业在多个方面做到精益求精。通过遵循相关法规、提交完善的申请资料、确保药品质量以及做好临床数据的准备,企业可以顺利通过审批,获得药品上市的通行证。对消费者来说,国药准字号是确保药品安全有效的有力保障,而对药企来说,成功申请国药准字号则意味着产品可以进入广阔的市场,满足更多患者的需求。