在中国,药品的销售和流通必须获得国家药品监督管理局(NMPA)批准的“国药准字号”证书。这个准字号是药品合法上市的前提,它意味着该药品符合国家药品的安全性、有效性以及质量的要求。随着中国医药市场的不断扩大和政策的逐步完善,越来越多的国内外药品企业纷纷向中国市场进军。申请国药准字号的条件到底是什么?本文将为您详细解析。
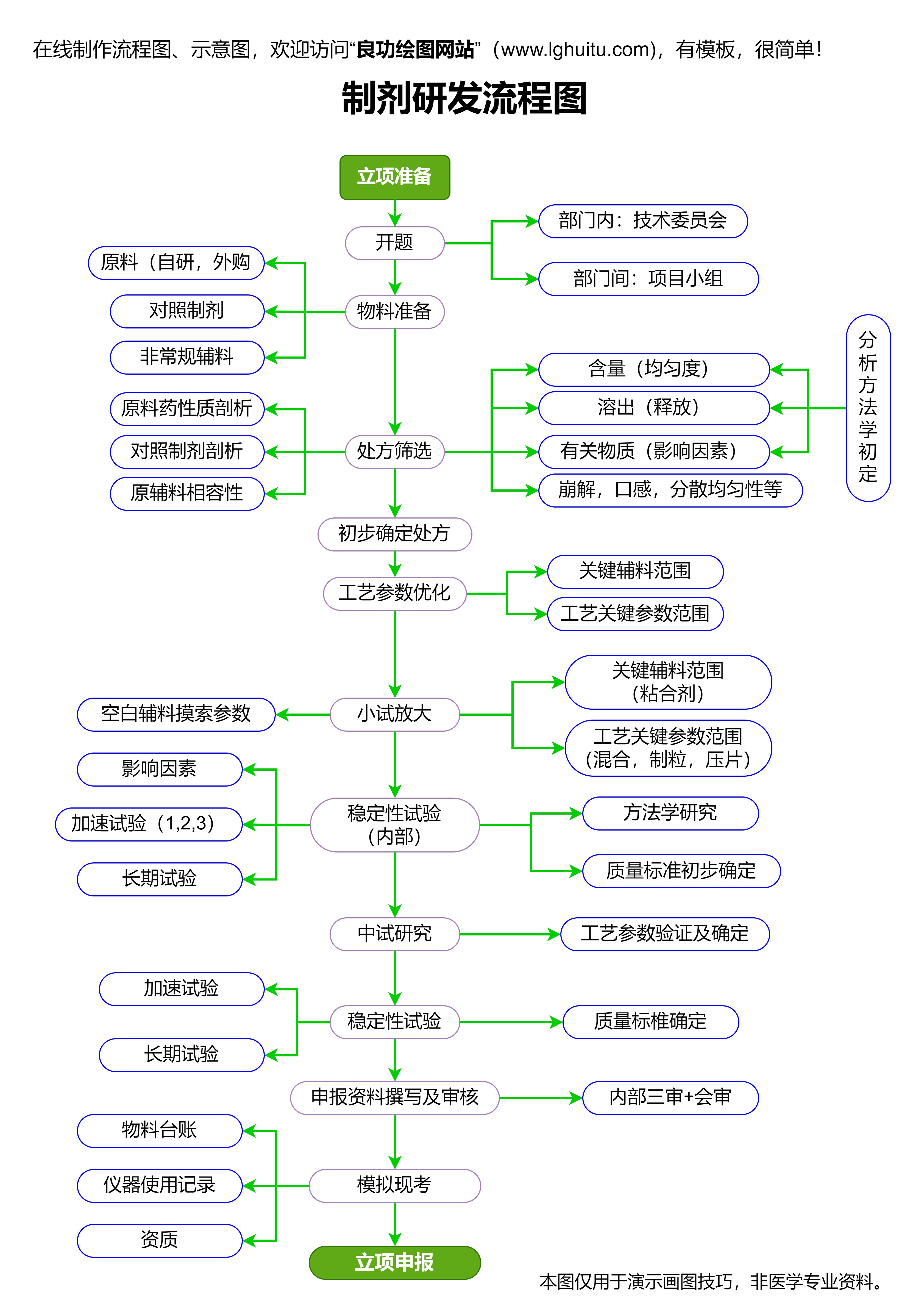
申请国药准字号的基础要求之一是药品的研发必须经过严格的科学论证和临床验证。药品研发不仅仅是理论上的构思,更需要在实验室和临床试验中进行大量的实验数据收集,确保药品的安全性和有效性。特别是针对新药品的研发,申请者必须提供足够的临床数据,证明药品能在临床应用中实现预期的疗效并且不产生重大副作用。
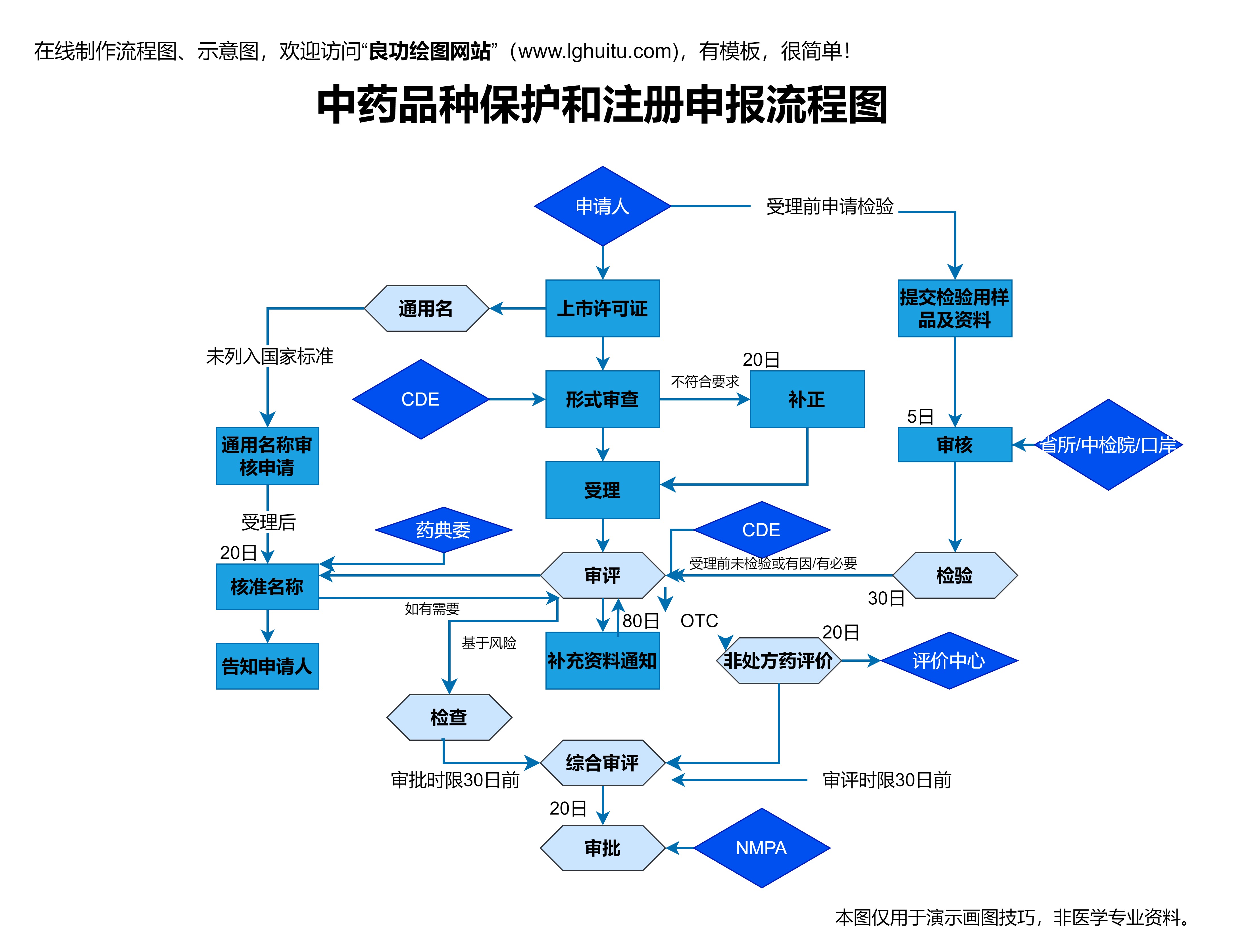
申请国药准字号的药品必须满足中国药典以及相关国家法规的要求。中国药典作为国家药品标准的官方文件,详细规定了药品的各项质量要求,包括原料药的质量、药品的生产工艺、质量控制方法等。申请国药准字号时,药品生产商需确保其产品符合中国药典的相关标准。如果是国外药品,生产企业还需提供符合中国药品法规的合规证明。
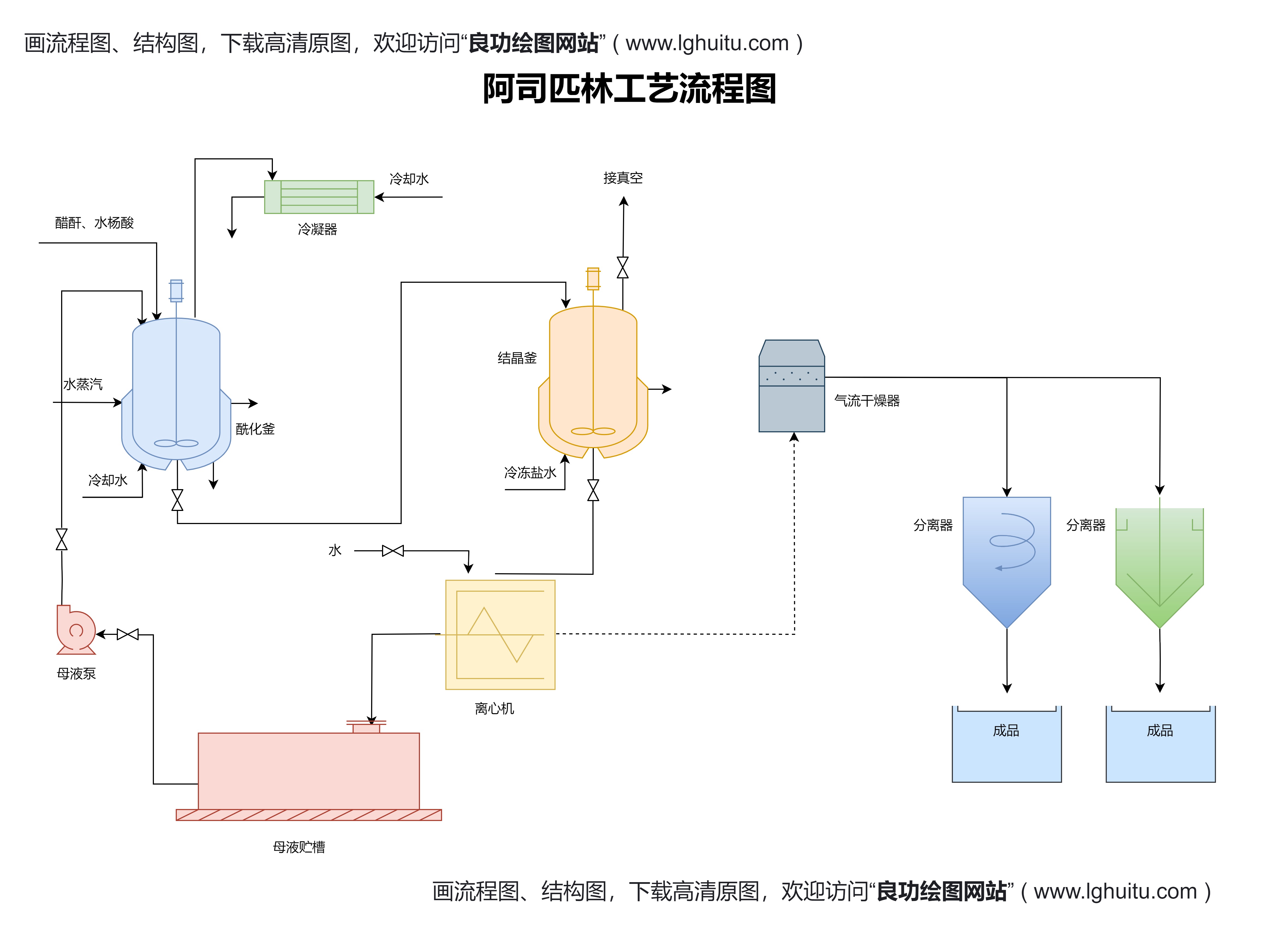
除了药品本身的质量要求外,生产企业的资质也非常重要。申请国药准字号的企业需具备符合条件的GMP(药品生产质量管理规范)认证。GMP认证是药品生产质量管理的核心标准,代表了企业在药品生产过程中的质量控制水平。无论是国内企业还是国外药品企业,只有经过严格GMP认证的生产设施,才能获得生产和销售药品的资格。对于国外药品企业,必须证明其生产的药品符合中国的GMP标准,或接受中国相关部门的检查和评估。
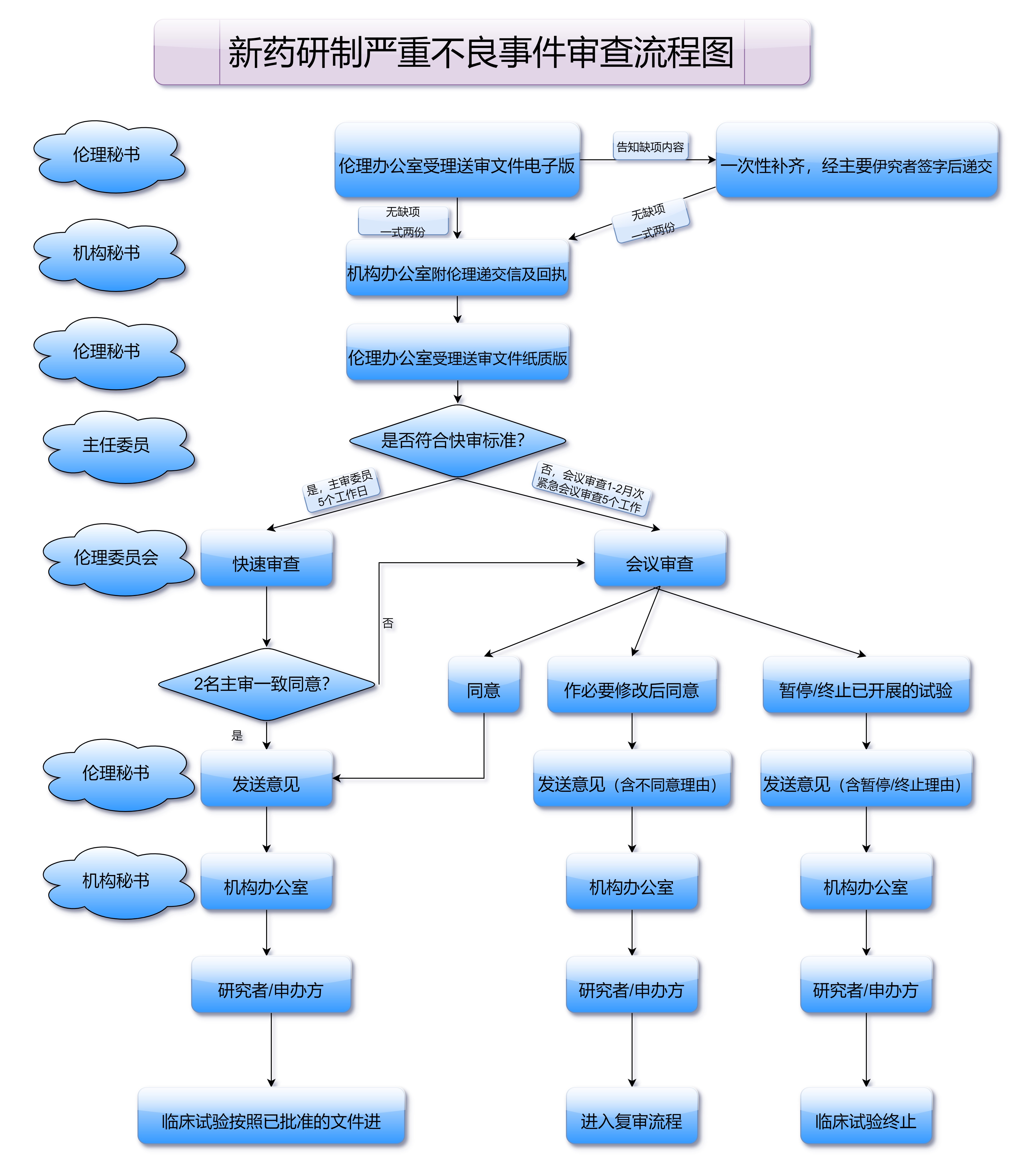
药品的包装和标签也必须符合国家药品法规的要求。申请国药准字号时,药品包装上必须清晰标注药品名称、生产厂家、有效期、批号、成分含量、用法用量等信息,确保消费者能够准确了解药品的使用方法和注意事项。尤其是在药品包装上,必须明确标注中文说明书,方便消费者使用。

对于新药申请来说,除了满足上述基础条件外,还需要提供药品的非临床研究数据和临床试验数据。这些数据是药品是否能够获得审批的核心依据。申请者需要提交药品在动物实验和人体试验中的安全性与有效性数据,证明该药品对人体无明显毒副作用并能达到预期疗效。
药品的临床试验还需符合中国的临床试验管理规定,并且试验结果必须经过相关机构的审核。临床试验数据的完整性和真实可靠性是申请国药准字号的关键。因此,药品企业在进行临床试验时,必须严格按照国家药品监督管理局的要求,确保每一步试验的科学性和合规性。
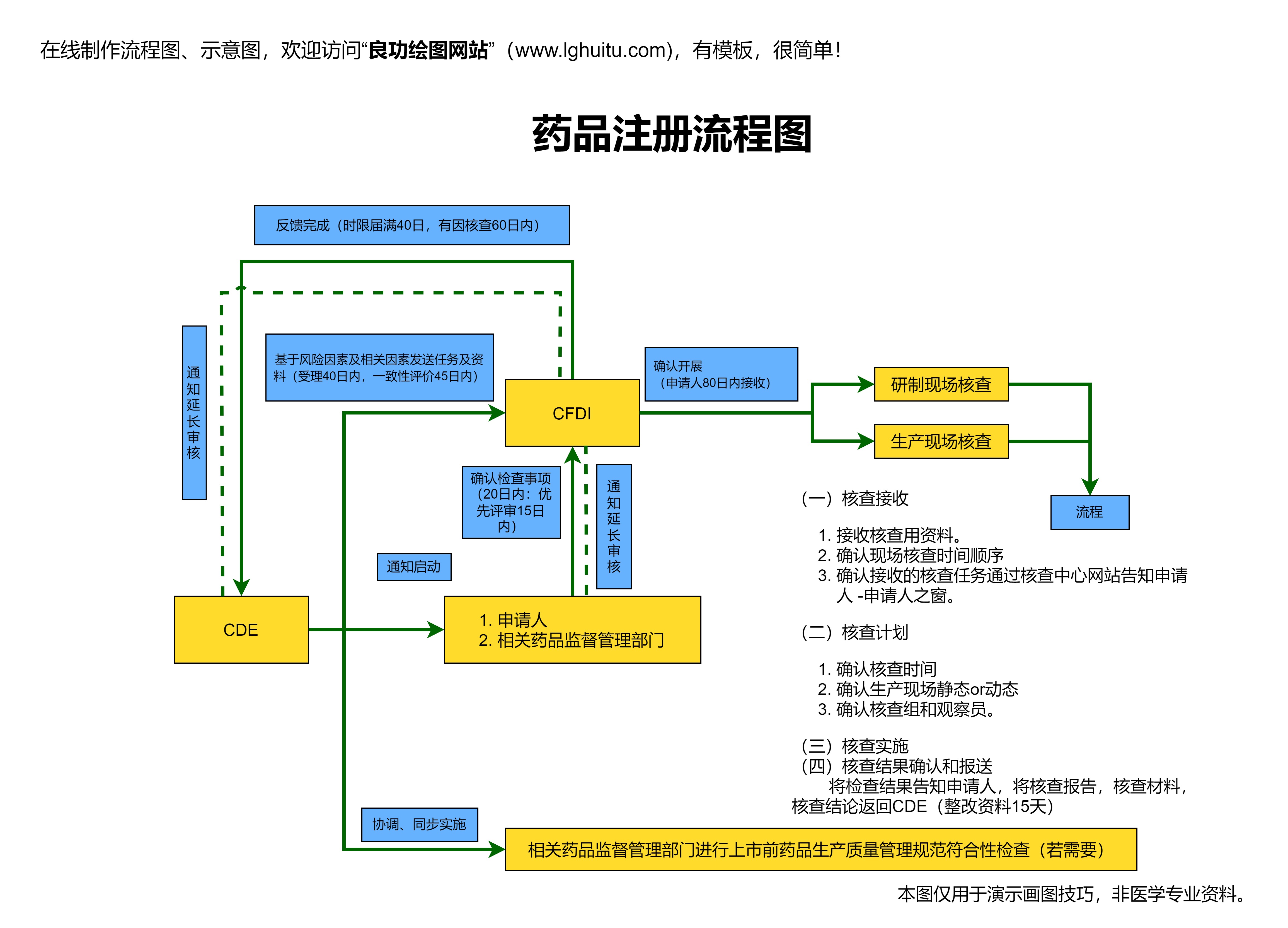
在申请过程中,申请者还需注意递交完整的资料和证明文件。申请药品的注册时,企业必须向国家药品监督管理局提交申请表、药品临床试验报告、药品生产和质量控制标准、药品注册申请资料等文件。申请过程中,企业还要确保所有资料真实、准确、完整,避免因资料不全或信息不准确而影响审批进度。
值得注意的是,药品审批的时间并非固定不变,申请国药准字号的审批过程通常需要数月甚至更长时间。企业在申请过程中应当与相关部门保持密切沟通,及时提供所需补充材料,确保顺利推进审批流程。对于已经获得临床试验许可的药品,企业还需提交临床试验完成报告,证明药品已通过必要的临床验证,并且能够满足市场需求。
药品的后续管理同样非常重要。即便药品获得了国药准字号,企业仍需进行定期的产品质量监督和安全性监测。药品上市后,生产企业必须及时向药监部门报告产品的质量情况、市场反馈以及任何不良反应。对于那些存在严重安全问题的药品,药监部门有权要求企业撤销销售或进行召回,以确保消费者的健康和安全。
中国药品市场的竞争日益激烈,因此,企业在申请国药准字号时,必须注重药品的质量、市场需求和品牌推广,确保药品不仅符合安全性和有效性标准,还能满足消费者的需求和期望。通过有效的市场调研、科学的研发投入和完善的质量管理,企业将能够在激烈的市场竞争中脱颖而出,赢得消费者的信任。
总结来说,申请国药准字号的条件十分严格,涉及药品的研发、生产、质量控制等多个环节。从申请资料的准备、临床试验的开展到生产工艺的合规性,企业需要付出大量的努力和资源。无论是国内企业还是国际药品公司,了解并遵守这些条件,将帮助他们顺利进入中国庞大的药品市场,抓住这一巨大机遇,实现商业成功。