药品研发是一个复杂且漫长的过程,涉及科学研究、临床实验以及严格的审批程序。从初步的药物筛选到最终的市场上市,每一阶段都需要经过严格的监管与审批。而新药的审批不仅是为了确保药物的安全性、有效性,更是为了保障公众的健康。为了更好地理解新药研发中的审批流程,我们将深入探讨从药物研发的初期阶段到临床试验的审批过程。
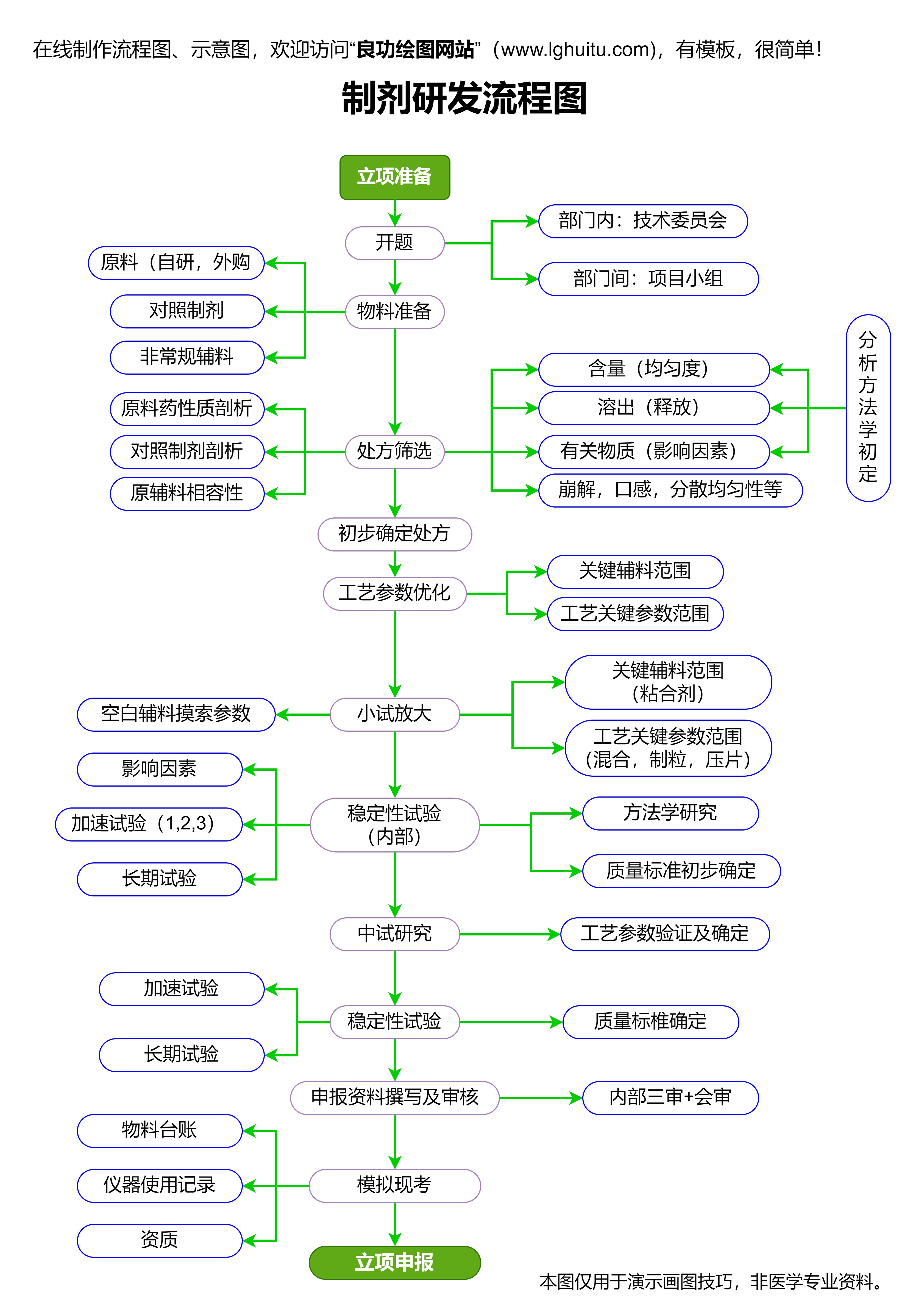
新药研发的第一步是药物的初步筛选。这一阶段通常由制药公司或科研机构的研究团队进行。研究人员会对潜在的治疗靶点进行探索,并筛选出具有可能治疗效果的化学分子、蛋白质或生物分子。初步筛选的药物分子需要通过体外实验和动物实验来评估其疗效和安全性。
一旦初步筛选出有效的药物候选分子,研发团队便会进入药理学研究阶段。这一阶段的重点是了解药物在生物体内的作用机制,包括药物的吸收、分布、代谢和排泄等过程。药物的初步安全性评估也是在这一阶段进行的,确保候选药物对人体不会产生明显的毒性。
药物研发的一个关键步骤是临床试验,这也是新药研发过程中审批的核心环节。临床试验分为多个阶段,包括临床前研究、临床I期试验、临床II期试验、临床III期试验和临床IV期试验。
临床前研究是指在人体临床试验前进行的实验研究。它主要包括动物实验和体外实验,旨在评估药物候选分子的安全性、毒性、药效和药代动力学特性。通过这些研究,研发团队能够了解药物在不同剂量下对生物体的影响,为后续的临床试验做好充分准备。
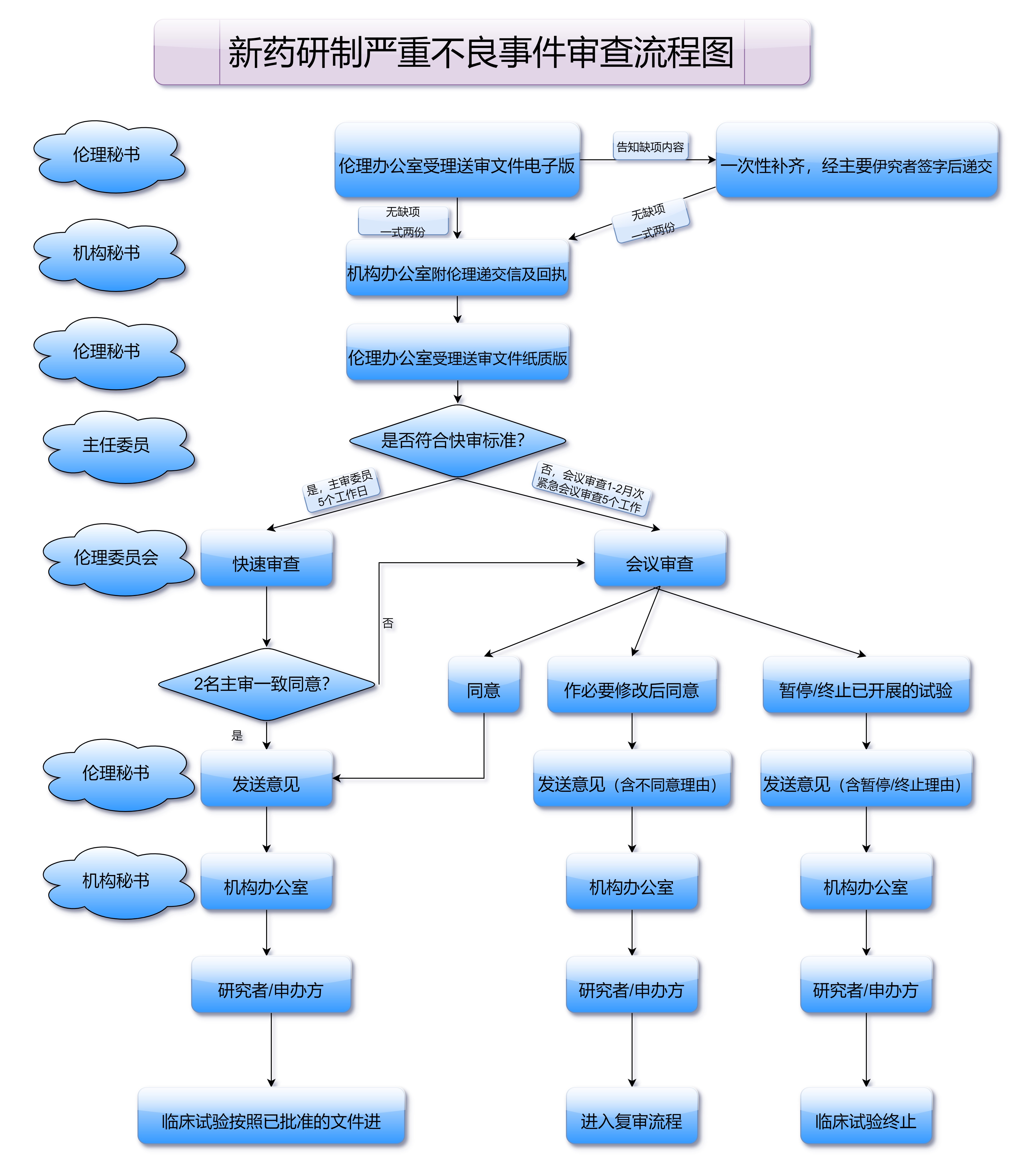
临床前研究的审批通常由国家药品监督管理部门(如中国国家药品监督管理局,简称NMPA)进行。药品研发公司需向监管机构提交药物的临床前研究报告和相关数据,以申请临床试验的批准。
临床I期试验是新药研发过程中的第一步临床试验。该阶段的主要目标是评估药物的安全性和耐受性。通常,临床I期试验只会招募少量健康志愿者,研究药物的剂量、使用方式和可能的副作用。通过I期试验,研究人员能够确定药物的安全剂量范围,并为后续的临床研究奠定基础。

在I期试验阶段,监管部门会对药品的审批严格把关,确保参与试验的志愿者不会面临过大的安全风险。通过临床I期的审批,药品研发团队获得了开展下一阶段临床试验的许可。
临床II期试验旨在评估药物的疗效和进一步评估其安全性。在这一阶段,药物将被应用于患有目标疾病的患者身上,研究其治疗效果和副作用。II期试验通常涉及数十到数百名患者,研究者会根据患者的反应来调整药物的剂量。
II期试验的审批通常比I期试验更为复杂,因为它不仅要关注药物的安全性,还要考察药物的疗效。监管部门会要求药品研发公司提交详尽的试验数据,并根据这些数据决定是否允许药物进入III期试验阶段。
临床III期试验是新药研发中最为关键的一步。该阶段的目标是全面验证药物的疗效和安全性,通常涉及数百到数千名患者。III期试验的结果直接影响药物是否能够获得上市批准。在这一阶段,研究人员会通过随机对照试验(RCT)等科学方法,确保药物相较于现有治疗方法具有显著的治疗效果。
III期试验的审批尤为严格,监管部门会要求提供大量的临床数据,以评估药物在实际应用中的效果和风险。这一阶段的成功不仅是新药上市的重要依据,还能够决定药品在市场上的竞争力。
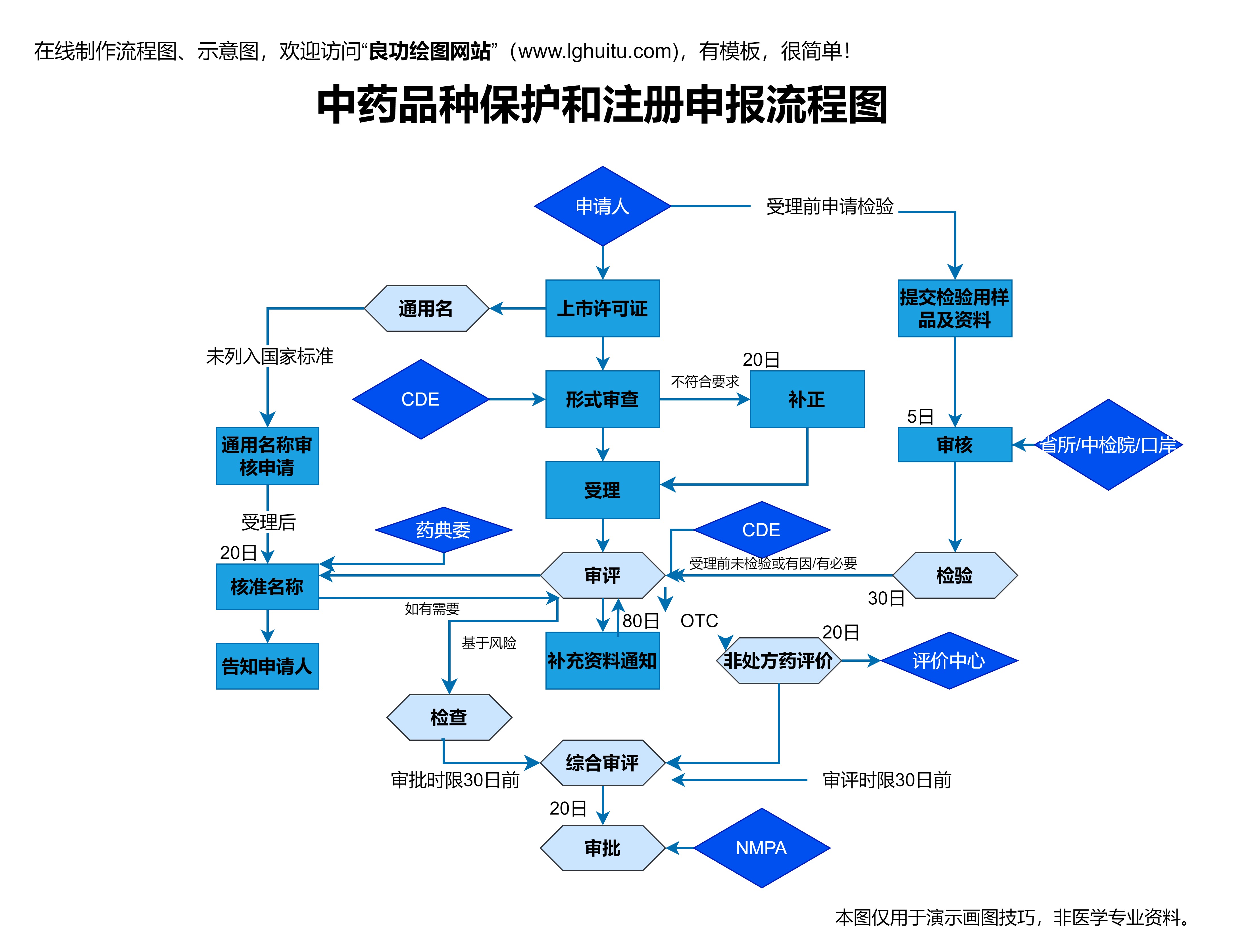
在新药研发过程中,国家药品监管部门起着至关重要的作用。在中国,国家药品监督管理局(NMPA)负责对新药的审批进行全面监管。NMPA对新药的审批过程进行严格把关,以确保药品的安全性、有效性和质量。
在药品研发的不同阶段,NMPA会根据药品的临床试验数据进行评估,并为药品进入下一个阶段提供必要的审批。这一多层次的监管系统确保了新药研发的高标准和高质量,能够为公众提供更安全、更有效的药物。
新药研发成功后,虽然已经经历了长时间的临床试验和严格的审批流程,但药品仍需经过一系列的审批程序才能正式进入市场。药品的上市审批涉及多个环节,其中包括药品注册、市场监测以及药品的定期评估和再审批。药品上市后的监管也是确保药物长期安全和有效的关键环节。
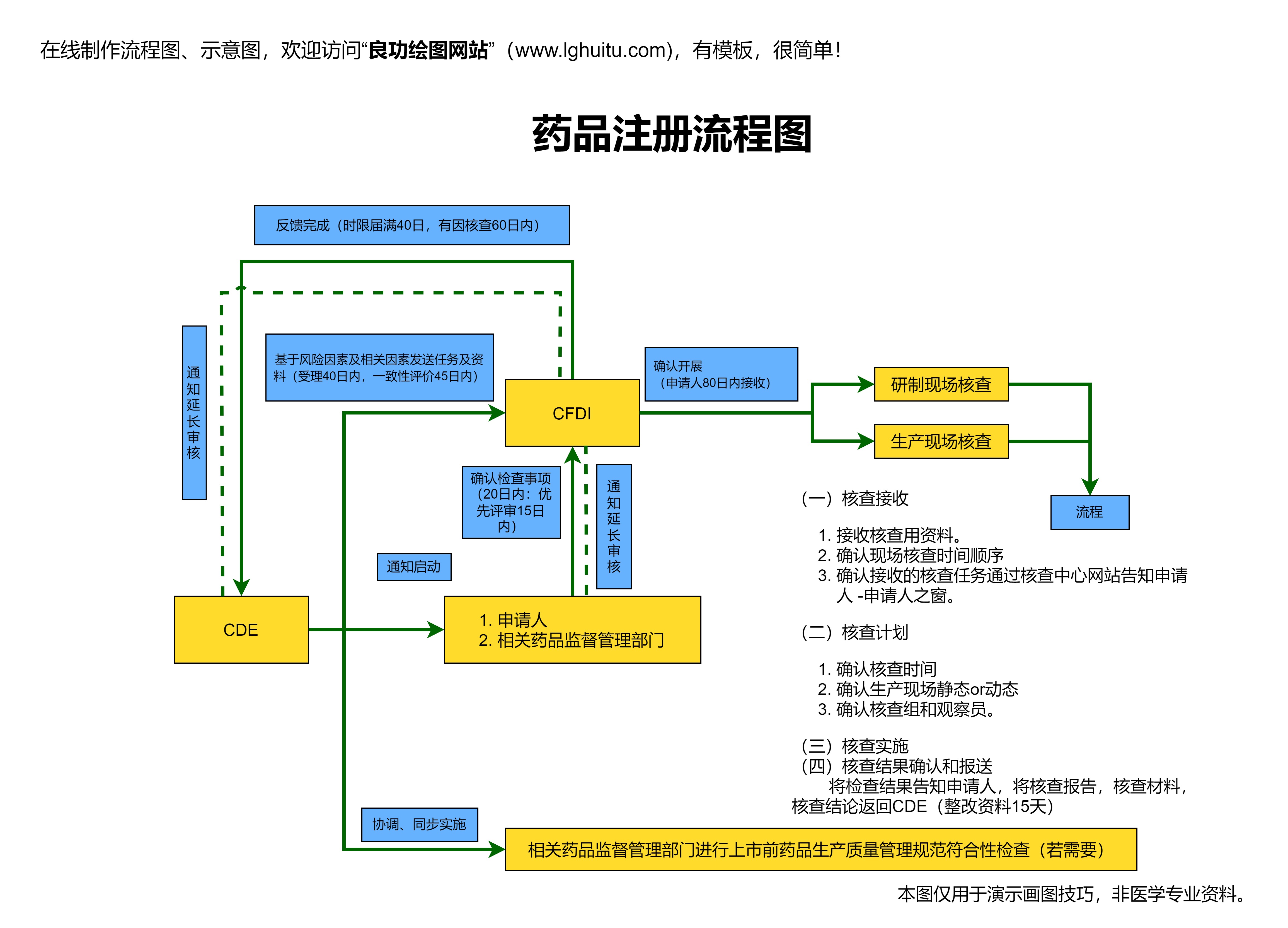
药品注册是新药上市的最后一步。药品研发公司在完成所有临床试验后,需向国家药品监管部门提交药品的注册申请,申请中包括药物的生产工艺、质量控制标准、临床试验数据等详细信息。监管部门会根据提交的材料,对药物的疗效、安全性、生产工艺等进行全面评估,决定是否批准药品上市。
药品注册的审批是一个复杂且高标准的过程,监管机构会审查药品的所有相关资料,并可能要求研发公司补充更多的研究数据。药品注册的审批还涉及到对药物生产设施的审查,以确保药品在生产过程中符合国家的质量标准。
药品上市后,仍需接受监管部门的持续监测。这一过程被称为药品的上市后监管。上市后监管主要包括药品的不良反应监测、药品的生产质量控制、市场的销售情况等。监管机构会定期对药品的使用情况进行评估,以确保药品在市场上的持续安全性。
尤其是对于一些新的治疗领域或高风险药物,监管部门会要求药品研发公司进行更严格的上市后监测。这些监测数据将作为药品长期使用和定期再审批的依据。
在药品上市后,监管部门还会进行定期的再审批。药品的定期再审批通常依据药品上市后的临床效果和不良反应数据进行评估。如果药品在市场上使用过程中出现了严重的安全问题或疗效不如预期,监管部门可能会要求药品进行重新评估,甚至暂停其销售。
再审批机制的目的是确保药品在长期使用过程中始终保持其安全性和有效性。通过这一机制,监管部门能够及时发现潜在的风险,并采取必要的措施保护公众健康。
从药物的研发到上市,每一步都涉及严格的审批和监管,确保每一种新药在上市前都能经过充分的科学验证和临床试验。而药品上市后,监管部门还会持续进行监测和评估,确保药物在长期使用中保持其安全性和有效性。这一系列的审批和监管流程体现了国家对公众健康的重视,同时也保障了新药研发过程中每一环节的合规性和高标准。
在未来,新药研发和审批的流程可能会随着科学技术的进步和药品监管政策的变化而不断优化。无论如何,药品的安全性和疗效始终是新药研发和审批过程中最重要的标准。