在当今的制药行业,药品的研发和上市是一个充满挑战的过程。药品注册审批流程是每一款新药能够进入市场的关键环节。随着国家对药品审批标准和监管的日益严格,企业必须在这一流程中精益求精,以确保产品符合所有的法律法规和安全标准。药品注册审批流程到底是怎样的?对于制药企业来说,如何能够顺利地通过这一流程呢?
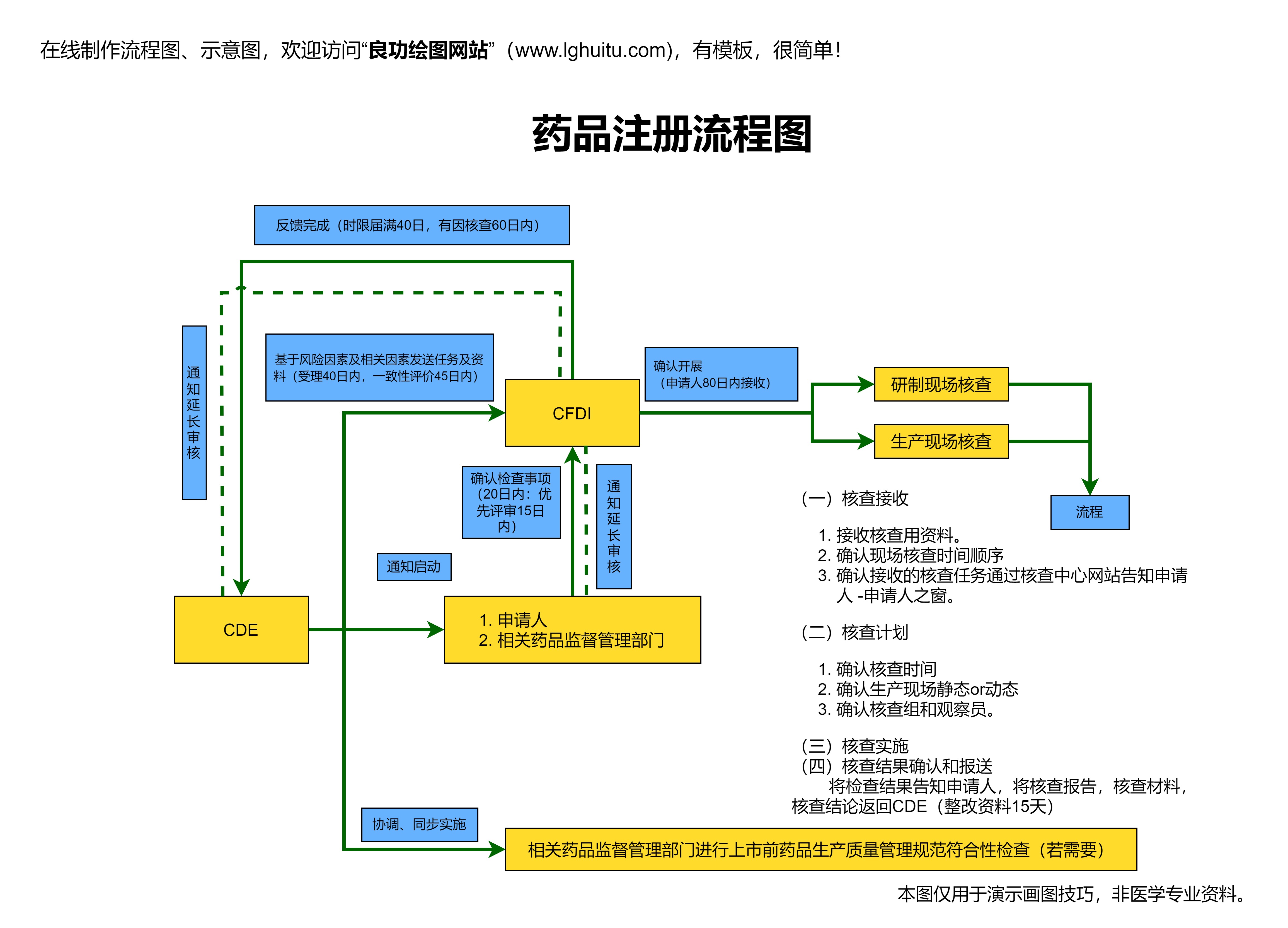
药品注册审批流程并不是一成不变的,它受制于国家的法律法规和药品监管政策。中国药品注册的审批流程较为严格,涉及多个环节,且每个环节都需要详细的资料和完善的操作。整个流程通常包括药品研发阶段、临床试验阶段、药品注册申请阶段、以及审批与上市阶段四大部分。
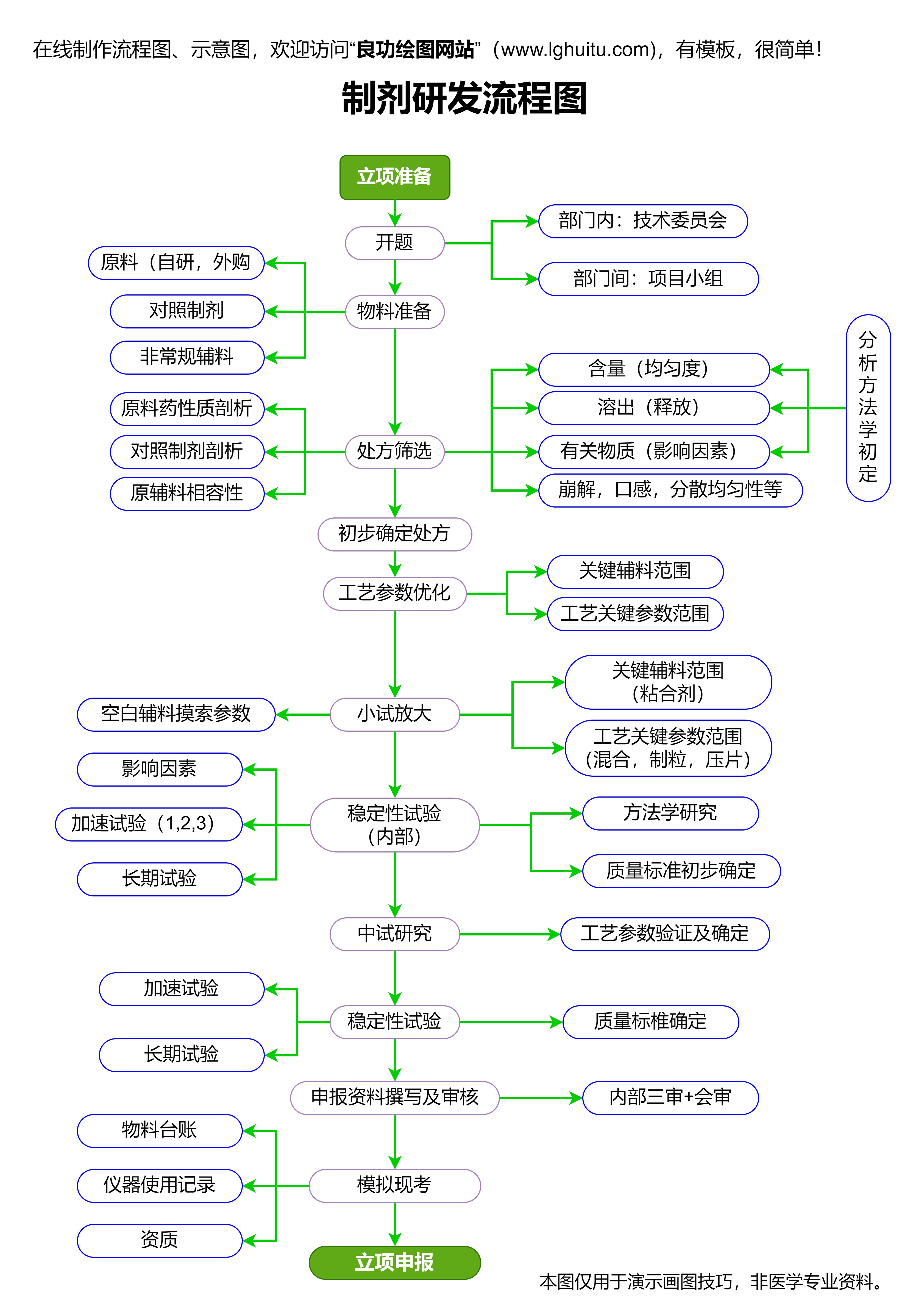
药品研发是整个药品注册过程中的第一步。在这一阶段,研发人员需要对药物的治疗作用、剂型、药理学、毒理学等方面进行全面研究。研究不仅要符合国际规范,还必须符合国内药品的相关标准。这一阶段非常重要,因为它为临床试验和药品注册申请提供了理论依据和基础数据。
在药品研发阶段,企业需要积累足够的科学证据,包括药物的化学成分、作用机制、临床前试验的结果等。这些资料在后续的药品注册审批中将会成为关键参考依据。
药品研发完成后,企业需要进行临床试验,评估药物在人类身上的安全性、有效性及药代动力学等各方面的表现。临床试验通常分为三期:Ⅰ期临床试验主要测试药物的安全性和耐受性;Ⅱ期临床试验则主要评估药物的疗效;Ⅲ期临床试验是药物的最终验证,目的是确认药物的疗效和安全性。
临床试验的数据收集和分析是药品注册中最为关键的一部分。只有通过严格的临床试验,才能证明药品的疗效和安全性,从而为注册申请提供有力支持。
药品完成临床试验并取得相关数据后,接下来便是药品注册申请阶段。在这一阶段,企业需要将所有研发数据、临床试验结果以及药品生产的质量控制标准等信息整理成一份详尽的申请材料。申请材料需要提交至国家药品监督管理局(NMPA),该局负责对药品的注册申请进行审查和审批。
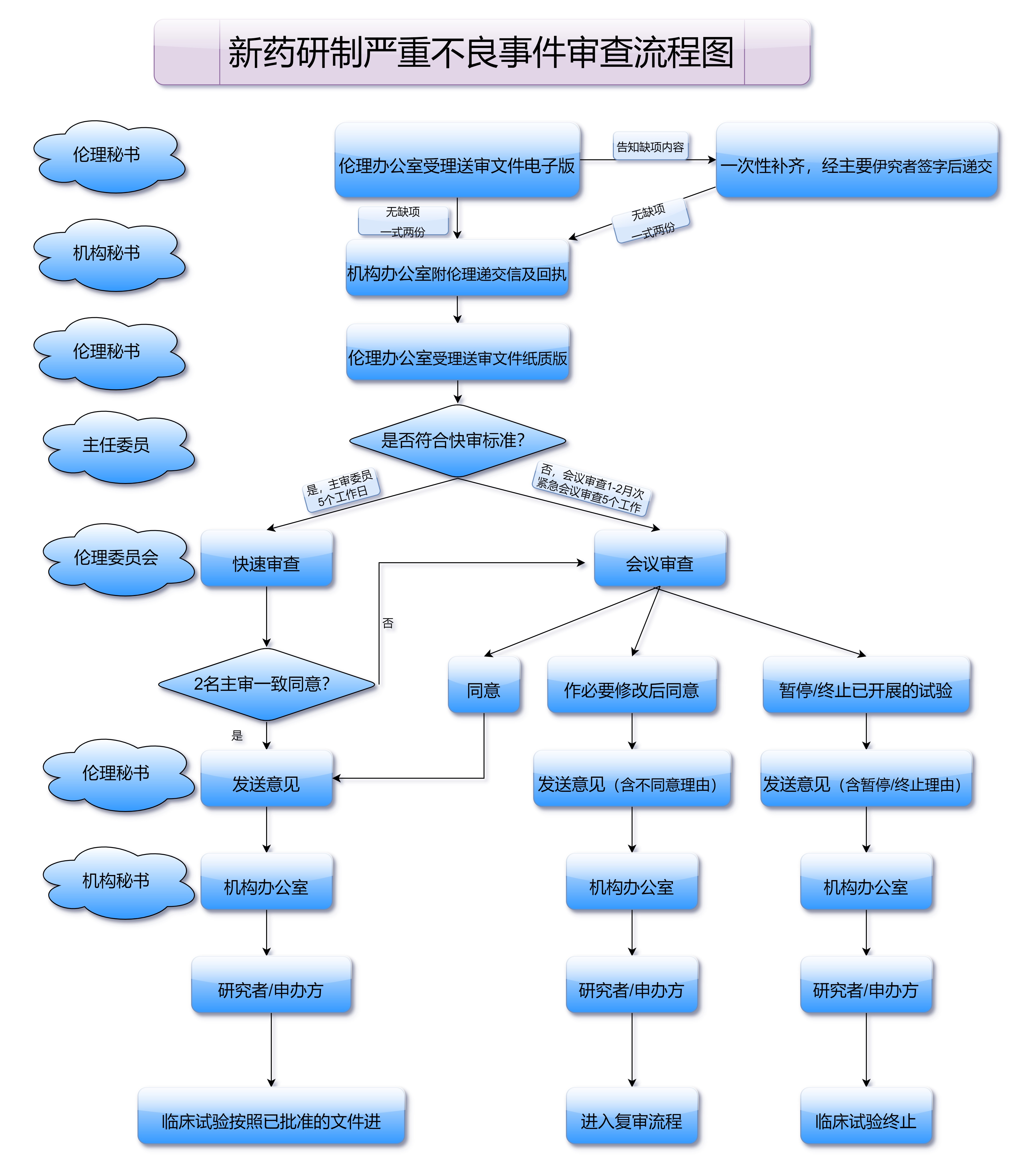
在药品注册申请的过程中,企业不仅需要提交完整的资料,还要确保所有文件符合NMPA的要求,并且没有遗漏。一个小小的细节失误都有可能导致审批延误,甚至无法获得批准。因此,在此阶段,企业通常需要与专业的药品注册机构和律师团队进行合作,确保注册申请的顺利进行。

在药品注册申请提交后,国家药品监督管理局将对申请材料进行严格审查。这一审查过程通常分为初步审查和专业审查两个阶段。初步审查主要检查提交的材料是否完整,是否符合格式要求。如果初审通过,进入专业审查阶段,专业审查会深入研究药物的疗效、安全性以及生产质量控制等方面。
在专业审查过程中,NMPA会要求提交更多的补充材料,或者要求企业对某些数据进行进一步验证和修正。这个过程可能需要数月甚至更长的时间。对于药品研发企业来说,耐心和细致的工作态度至关重要。
一旦药品通过了NMPA的审批,企业就可以获得药品注册证书,并且可以开始在市场上销售该药品。不过,上市后药品仍然需要定期进行监管和跟踪,确保药品在使用过程中不会产生新的安全隐患。
尽管药品注册审批流程已经日益规范,但在实际操作中,企业仍然会面临一系列挑战。其中,最常见的问题包括:
资料不完整或不符合要求:药品注册过程中提交的资料需要详尽、准确,任何细节的疏忽都可能导致审批失败。企业必须在材料准备阶段投入充足的资源。
临床试验数据问题:临床试验是药品审批的核心,数据的真实性和准确性是审批能否顺利通过的关键。任何临床试验数据不合格,都可能导致审批被推迟或拒绝。
时间管理问题:药品注册过程复杂且周期较长,企业需要合理安排研发和审批时间,避免因延误导致药品上市错失市场机会。
药品注册审批流程是一个复杂且严格的过程,企业只有通过细致周到的准备,才能顺利通过每一个环节。随着药品市场竞争的加剧,企业需要更加注重药品注册的专业性和合规性,确保产品能够尽早进入市场,造福广大患者。
药品注册的每一个环节都需要精心策划和细致执行,只有全程把控每个细节,才能确保药品顺利通过审批,进入市场,实现企业的商业价值。