近年来,随着医疗科技的不断发展和药品行业的逐步创新,药品注册成为了制药企业开展业务的基础性步骤。为了确保药品的质量、安全性和疗效,国家药品监督管理局(NMPA)在2019年发布了《药品注册管理办法》,对药品注册的各个环节进行了详细的规范。该办法旨在进一步提升药品注册的透明度与效率,同时确保公众能够获得更为安全、有效的药品。

药品注册管理办法的实施意味着药品上市前的审批程序将变得更加严格。在过去,药品注册审批周期较长,过程繁琐。如今,通过优化审批流程和完善监管措施,药品的上市时间得到了有效缩短,药品质量得到了更加严格的保障。
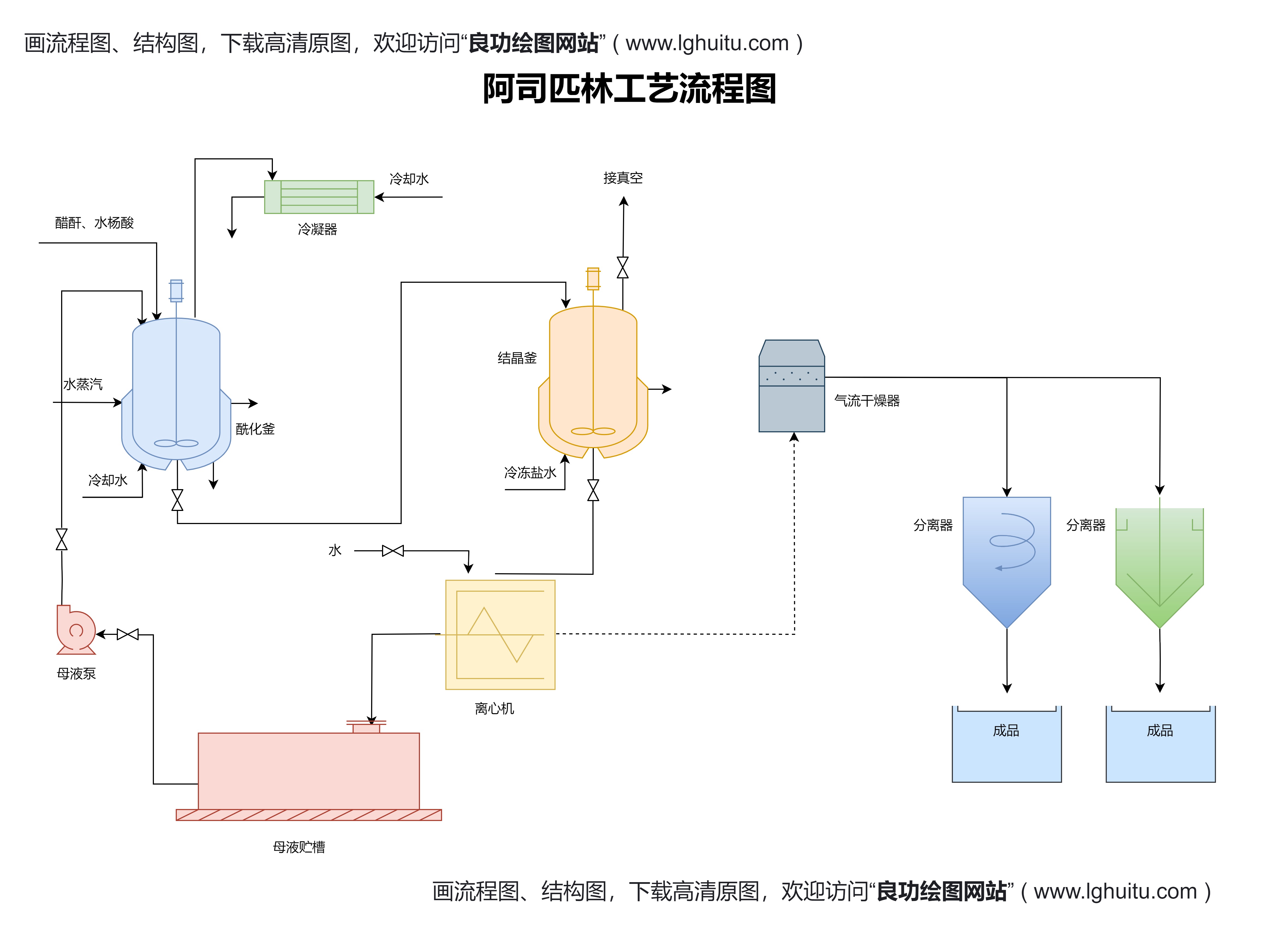
药品注册管理办法强调了对药品研发和生产环节的全过程监管。新的管理办法要求企业在药品研发阶段就开始向监管部门进行信息披露,确保药品的研发符合安全性和有效性的基本要求。对于生产环节,药品注册管理办法明确了生产质量控制的标准,企业必须按照规定的GMP(良好生产规范)进行生产,以确保药品的生产过程符合质量要求。
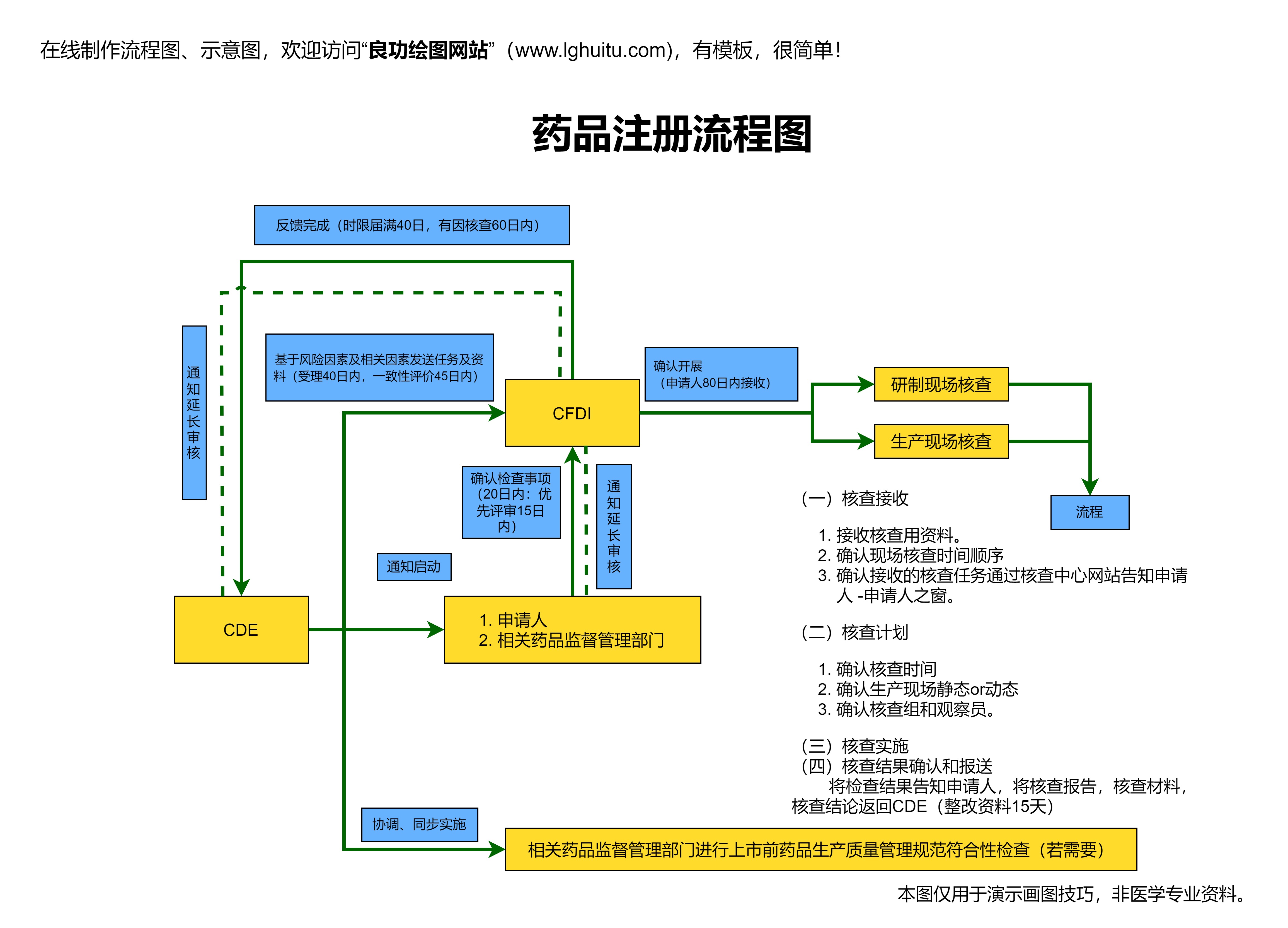
药品注册管理办法的核心目标,是确保药品能够以最合规、安全的方式进入市场,并为公众提供切实有效的健康保障。管理办法对药品的临床试验、生产质量、包装标签等方面均做出了明确规定。这些措施不仅增强了公众对药品质量的信任,也为制药企业提供了更为明确的指导。
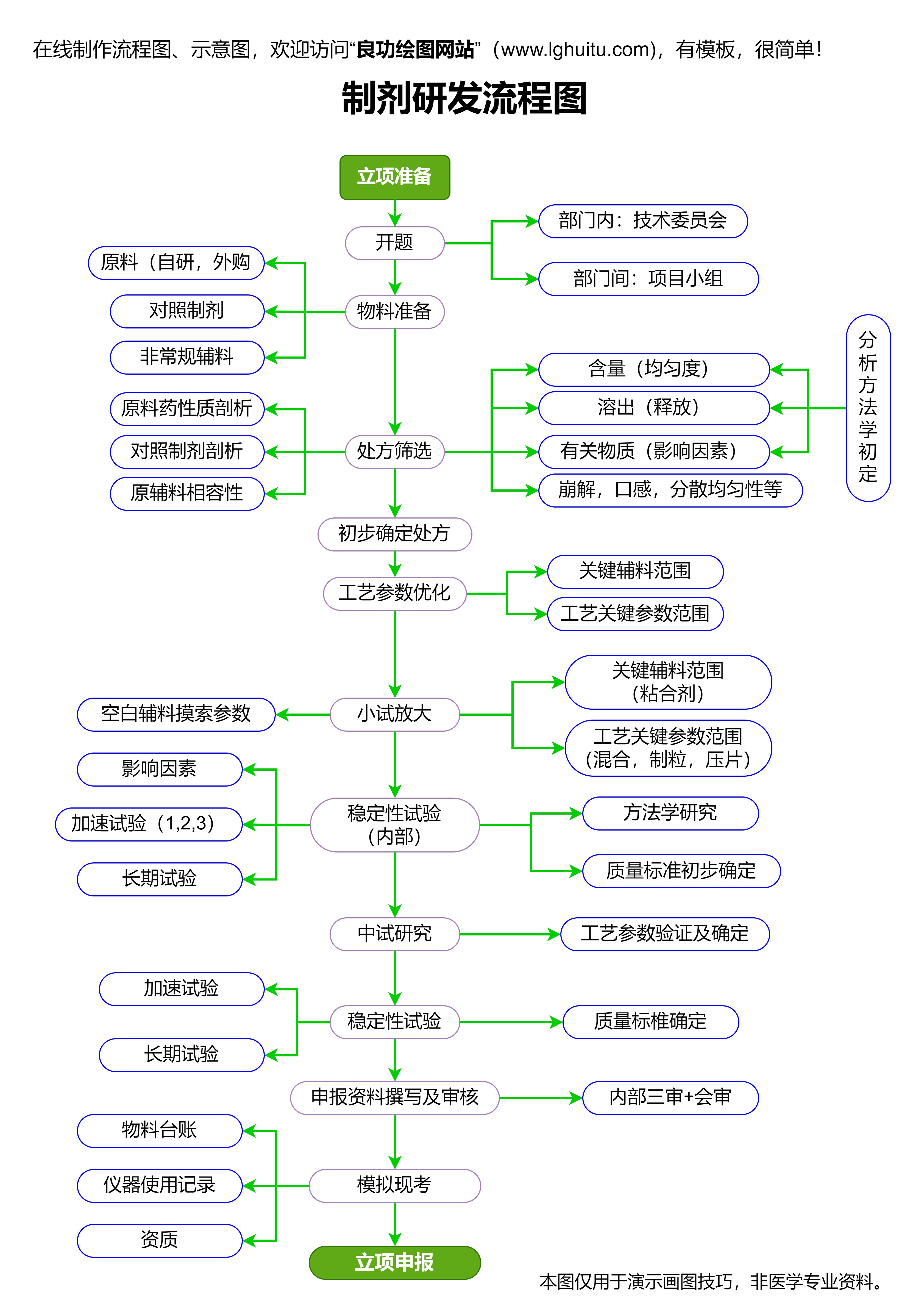
随着药品注册管理办法的逐步推进,行业内也对合规性提出了更高要求。对于制药企业来说,了解并准确掌握注册管理办法的实施细则,是顺利通过审批、顺利上市的关键。
药品注册管理办法的实施,对制药企业产生了深远的影响。它带来了审批流程的改革。新办法推动了药品注册的“分步审批”和“优先审批”机制,对于一些符合条件的创新药物,允许通过加快审批流程提前进入市场。通过这项措施,不仅加速了新药的上市进程,也为企业提供了更具竞争力的市场机会。
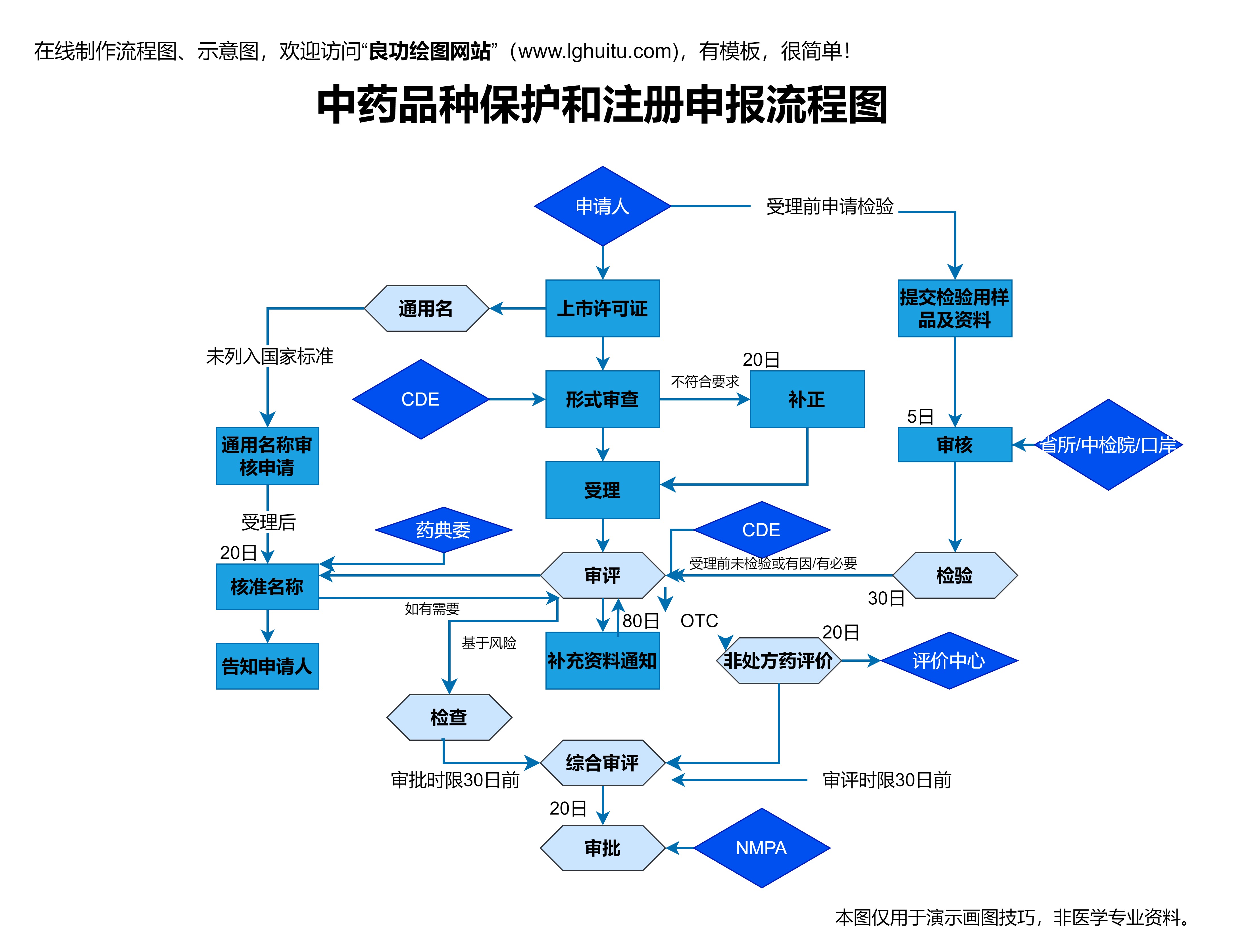
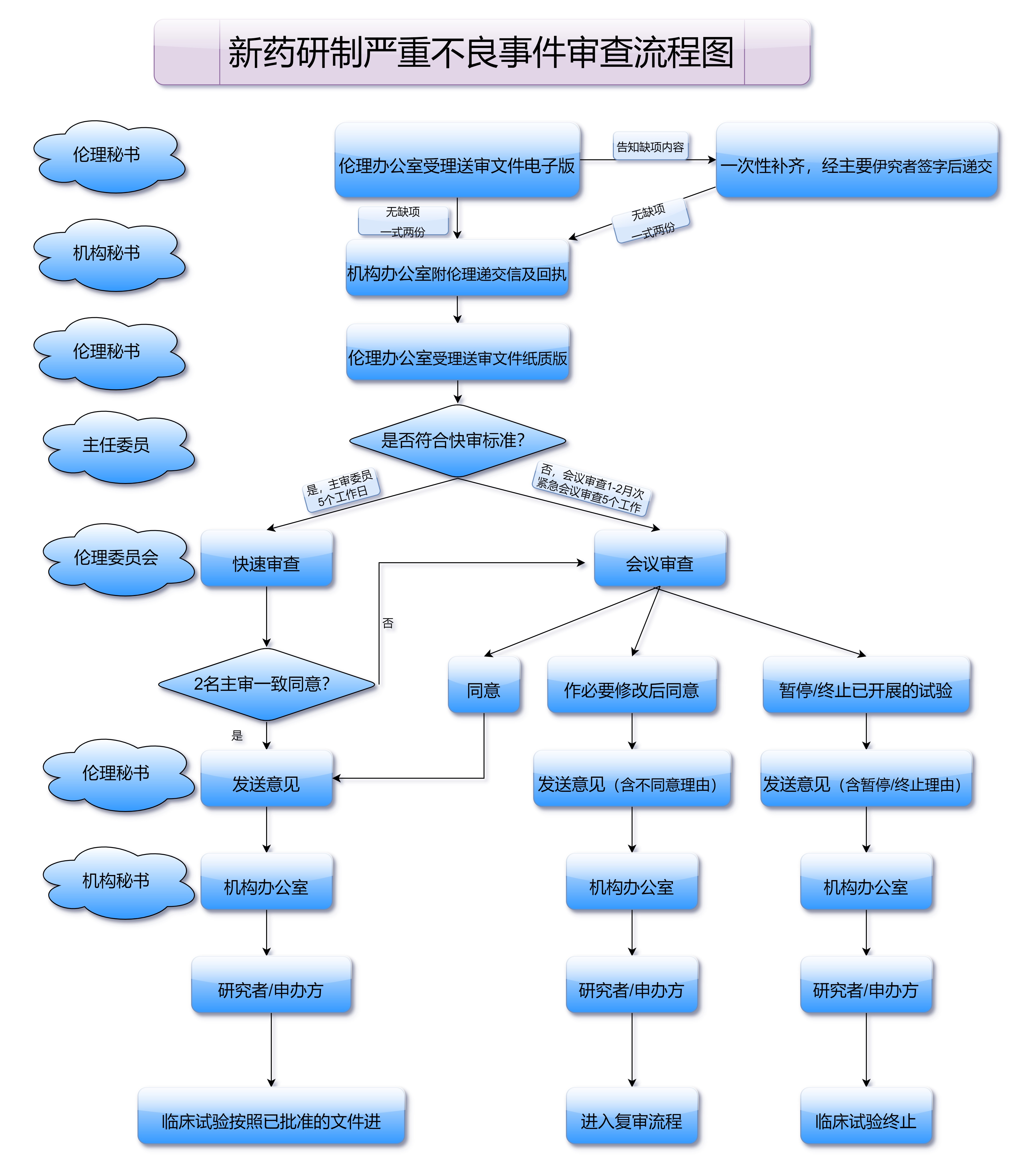
药品注册管理办法还对药品的临床试验提出了更高的要求。根据新规定,药品研发过程中必须提供充分的临床试验数据,并经过严格的审查。这一举措确保了药品的疗效和安全性,更好地保护了患者的健康。药品注册管理办法规定,药品临床试验必须符合GCP(临床试验质量管理规范),并要求药品在临床试验过程中确保科学性与伦理性的合规。
对于制药企业而言,如何应对药品注册管理办法的实施,成为了其成功获得药品上市批准的关键。企业需要加强与监管部门的沟通,确保每一个药品的注册申请都能符合政策要求。在注册过程中,企业应做好全程的资料整理与数据审查工作,确保提交的资料完整、真实和准确。
企业应注重研发投入,尤其是在药品安全性、有效性和质量控制方面。随着国家对药品质量的要求逐步提高,制药企业必须投入更多的资金和资源,提升研发技术和生产能力,确保药品能够符合注册要求。
药品注册管理办法不仅提高了药品的质量和安全性,也促进了制药行业的规范化发展。面对新的注册管理办法,企业应主动应对,优化研发和生产流程,以更高的标准要求自己,确保能够顺利完成药品注册,进入市场,最终为患者提供更加安全和有效的治疗选择。