在医疗器械行业中,了解医疗器械的分类标准至关重要。根据我国《医疗器械监督管理条例》以及相关规定,医疗器械被分为1类、2类、3类三类。每类器械的管理规定、注册要求以及审批程序都有显著区别。不同类别的医疗器械涉及到的风险等级不同,管理的严格程度也相应提升。这一分类制度旨在确保医疗器械的安全性与有效性,保障公众健康。
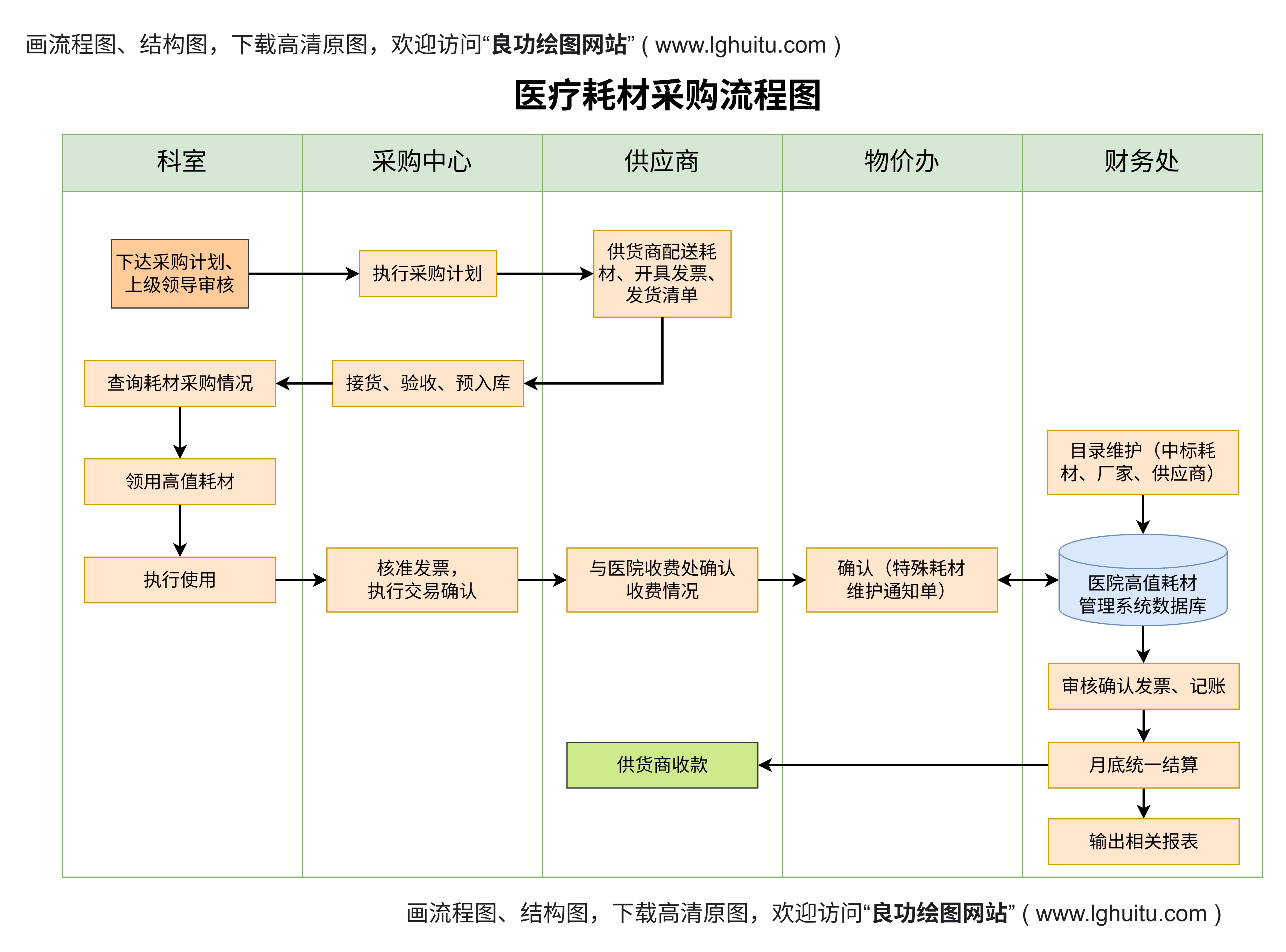
1类医疗器械指的是那些风险较低,且基本上不涉及复杂技术的器械。这类器械通常不对人体产生重大影响,因此相对来说,监管要求较为宽松。根据法规,1类医疗器械只需进行备案即可,不需要经过复杂的注册审批程序。比如,一些普通的医用手套、体温计、外用敷料等都属于1类医疗器械。

对于生产和销售1类医疗器械的企业来说,最重要的是确保产品符合国家的基本安全和卫生要求,且生产工艺应符合相关的质量管理标准。虽然1类医疗器械不需要进行繁琐的注册审批,但企业仍需保持良好的生产记录和质量管理体系,确保产品持续符合标准要求。

2类医疗器械的风险程度较1类器械稍高,因此需要进行更为严格的注册审批。对于这一类器械,企业不仅需要进行产品的备案,还需要提供相应的临床试验数据、质量管理体系认证等证明文件。这类器械通常涉及一定的技术复杂性或对人体有一定的影响。例如,超声诊断设备、输液器、血糖仪等都属于2类医疗器械。
2类医疗器械的监管要求较为严格,审批周期较长,因此企业在进行产品设计、开发时,需要充分考虑法规要求,确保产品符合各项标准。除了必要的注册程序外,企业还需要进行定期的产品检验、生产现场检查等,以确保产品的持续合规性。
3类医疗器械是风险等级最高的一类,通常涉及对人体健康有重大影响的器械,且其技术复杂度高,使用时可能产生较大的风险。例如,植入性器械、心脏起搏器、人工关节等均属于3类医疗器械。
这类器械的管理要求最为严格,企业在申请注册时需要提供详细的临床数据、动物实验结果及产品的安全性与有效性分析报告。3类医疗器械的审批过程不仅复杂且周期较长,需要经过多项审批程序。在产品投放市场之前,企业还需对产品进行长时间的跟踪监测,以确保其在实际使用中不会出现不良反应。
由于3类医疗器械涉及较大的风险,企业需要有完善的研发和生产体系,确保产品符合国际标准和国内法规要求。企业还需要关注产品上市后的监管,及时响应可能出现的质量问题或风险。
了解医疗器械的分类标准是企业进行市场布局、产品研发和合规管理的前提。不同类别的医疗器械不仅在设计和生产阶段有不同的要求,上市后的监管与维护也有所不同。企业在应对这些要求时,需要注重以下几点:
医疗器械的风险管理要求是分类标准中的重要一环。无论是1类、2类还是3类医疗器械,企业都需依据不同的风险等级采取相应的风险控制措施。对于1类医疗器械,企业应确保产品设计简单且无重大风险;对于2类和3类医疗器械,企业则需进行更为详细的风险评估,确保产品在使用中的安全性和有效性。
特别是2类和3类医疗器械,企业需要建立完善的风险管理体系,包括产品的设计验证、质量控制、临床试验以及上市后跟踪等。只有这样,才能确保产品在投入市场后长期稳定,避免出现不合规或安全隐患。
医疗器械的注册与审批流程是企业进入市场的关键步骤。不同类别的医疗器械有不同的注册要求,企业必须根据产品的风险等级和技术特点,提前做好相关准备工作。1类医疗器械的备案流程相对简单,但对于2类和3类医疗器械,企业需要提前准备好临床数据、实验报告、质量管理文件等,确保能顺利通过注册审批。
在准备材料时,企业要注重与相关监管部门的沟通,确保所有文档符合要求。要根据不同市场的法规要求,及时调整注册策略,以便顺利进入目标市场。
无论是哪类医疗器械,企业都必须建立健全的质量管理体系,确保从研发、生产到销售的每一个环节都符合国家标准与国际质量要求。尤其是对于2类和3类医疗器械,企业还需要进行定期的质量评估与审计,确保产品在使用过程中的安全性。
随着产品上市后可能会出现新的安全风险或技术问题,企业应建立完善的产品监测和反馈机制,及时响应市场上的不良反应。通过后期的持续监管,企业不仅能够有效应对产品质量问题,还能在监管部门的审核中展示出良好的合规记录,提升企业信誉。
随着科技的进步,医疗器械行业的技术不断创新。企业在研发新产品时,除了要关注法规要求,还应借助先进技术提高产品的性能和安全性。通过科技创新,企业不仅能够满足日益严格的市场需求,还能提升产品的竞争力,为企业开辟新的市场空间。
未来,随着医疗器械市场的不断扩大,企业必须持续跟踪医疗器械分类标准的变化,确保产品的合规性。要注重技术创新,推动行业的持续发展,满足消费者对于健康和医疗的不断追求。
了解并遵守医疗器械分类标准对于企业的长远发展至关重要。通过对不同类别器械的管理要求进行全面掌握,企业能够更好地应对市场挑战,把握发展机遇,最终实现合规和创新的双重目标。