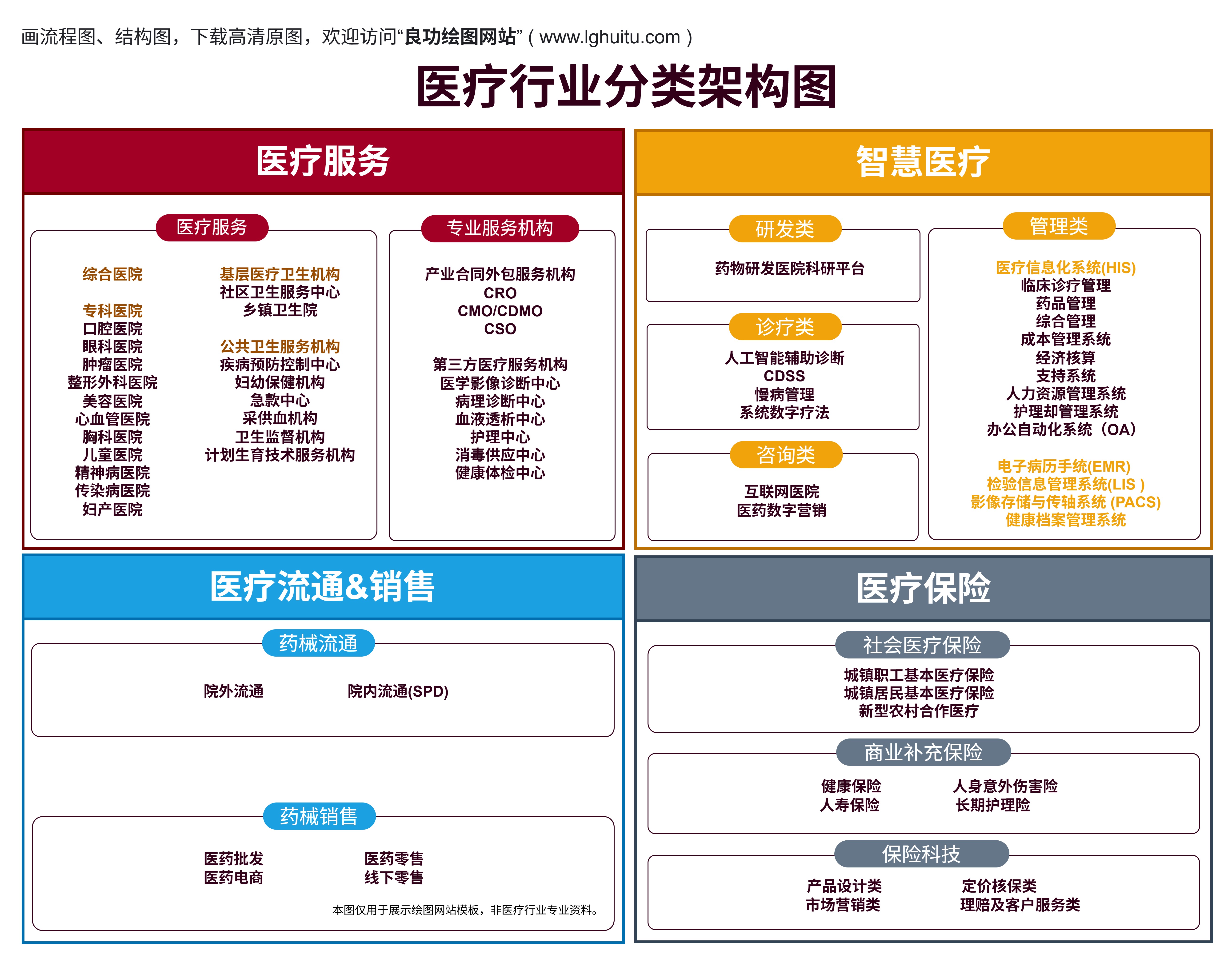
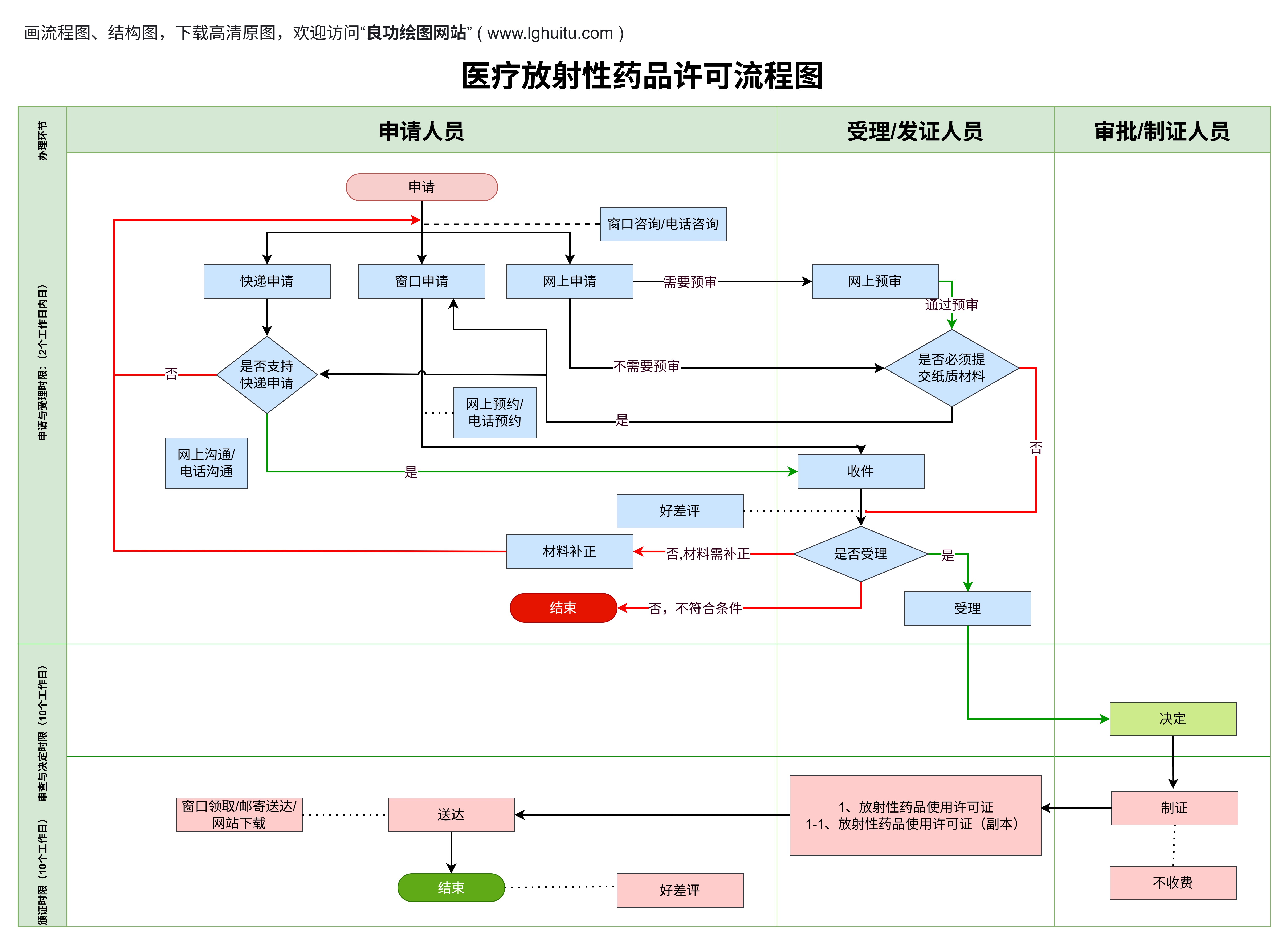
在现代医疗行业中,医疗器械的种类繁多,从简单的体温计到复杂的医疗影像设备,每一种器械都有不同的法规与资质要求。特别是在中国,医疗器械分为1类、2类和3类,每一类的资质要求和审批程序都有所不同。了解这些资质要求,是每一个医疗器械企业顺利进入市场的关键。本文将为您详细解析1类、2类和3类医疗器械所需的资质,帮助您清楚掌握如何满足相关法律法规的要求。

1类医疗器械是指风险较低的医疗器械,通常是那些不会对人体健康产生重大影响的产品。例如,手术器械、体温计、消毒剂等。1类医疗器械的特点是对人体健康的潜在风险较低,因此,其审批程序相对简单,所需的资质要求也较少。

在中国,1类医疗器械通常只需要向当地的食品药品监督管理局(简称:药监局)进行备案即可。这类器械不需要经过复杂的注册程序,但仍需要满足一些基本要求,比如:
产品质量管理体系:即使是1类器械,生产企业也需要建立符合质量管理要求的体系,确保生产过程符合规范,保障产品的安全性和有效性。
备案材料的提交:企业需向药监局提交包括产品说明书、技术文件、生产过程及质量控制等相关材料。药监局会对这些材料进行审核,确认产品符合安全标准。
标签和说明书要求:产品的标签必须明确标识产品类别、生产厂家、使用方法等信息,确保用户能够正确使用。
与1类医疗器械相比,2类医疗器械的风险稍高,通常涉及到对人体健康有一定影响的产品,如诊断设备、X光机等。2类医疗器械不仅仅需要备案,还需要经过药监局的注册程序。这意味着,生产和销售2类医疗器械的企业需要提供更加详细的产品资料,并经过严格的审核和批准。
产品注册申请书:申请注册时,企业需提供一份详细的注册申请书,介绍产品的功能、用途、设计及制造过程等信息。
临床试验数据:对于某些2类医疗器械,药监局可能要求企业提供临床试验数据,以证明产品的安全性和有效性。特别是对于新型器械或改进型产品,临床试验至关重要。
质量管理体系认证:类似于1类医疗器械,2类器械的生产企业也需要建立符合国际或国家标准的质量管理体系。这通常包括ISO13485等认证,以确保生产过程中的每个环节都能得到有效控制。
产品检测报告:企业还需要向药监局提交产品的检测报告,证明其符合相关的技术标准和安全要求。
通过注册和批准,2类医疗器械能够进入市场销售,但整个审批过程相对1类器械复杂,且审批周期较长。
3类医疗器械是指那些对人体健康具有较高风险的医疗器械,通常包括植入性设备、心脏起搏器、人工关节等。由于这些器械可能会直接影响到患者的生命安全,因此,3类医疗器械的资质要求和审批程序是最为严格的。
对于3类医疗器械,企业必须经过药监局的注册审批,且审批过程复杂且时间较长。3类医疗器械的资质要求主要包括:
注册申请书及技术资料:企业首先需要提交详细的注册申请书,其中包含产品的技术参数、设计理念、使用说明、生产工艺以及质量管理体系等资料。
临床试验与验证:对于3类医疗器械,药监局几乎总是要求提供临床试验数据。企业需要通过临床研究证明产品在实际应用中的安全性与有效性,并确保产品能够在临床环境中使用。
生产厂址及质量管理体系认证:与2类器械一样,3类医疗器械的生产企业需要获得ISO13485等国际质量管理体系认证,确保生产环境和产品符合全球最高质量标准。
产品检测与技术评估:对于3类医疗器械,药监局通常会要求提供多项独立的产品检测报告,包括生物相容性、安全性、稳定性等方面的技术评估,以确保产品在使用过程中不会对人体产生危害。
注册审核与批准:药监局在审核注册资料时,会特别关注产品的临床数据、风险评估报告以及质量管理体系。通过审核后,企业才能获得3类医疗器械的注册证书,并开始产品的生产和销售。
无论是1类、2类还是3类医疗器械,企业都需要遵循严格的法规要求,以确保产品的安全性、有效性和质量。对于不同类别的器械,资质要求也有所不同,审批程序也从简单到复杂不等。企业需要根据不同产品的特点,准备相关的申请资料,确保顺利通过药监局的审核,合法合规地进入市场。
如果您是医疗器械行业的从业者或计划进入这一行业,了解并熟悉这些资质要求,确保产品符合相关法规,将是您成功的第一步。