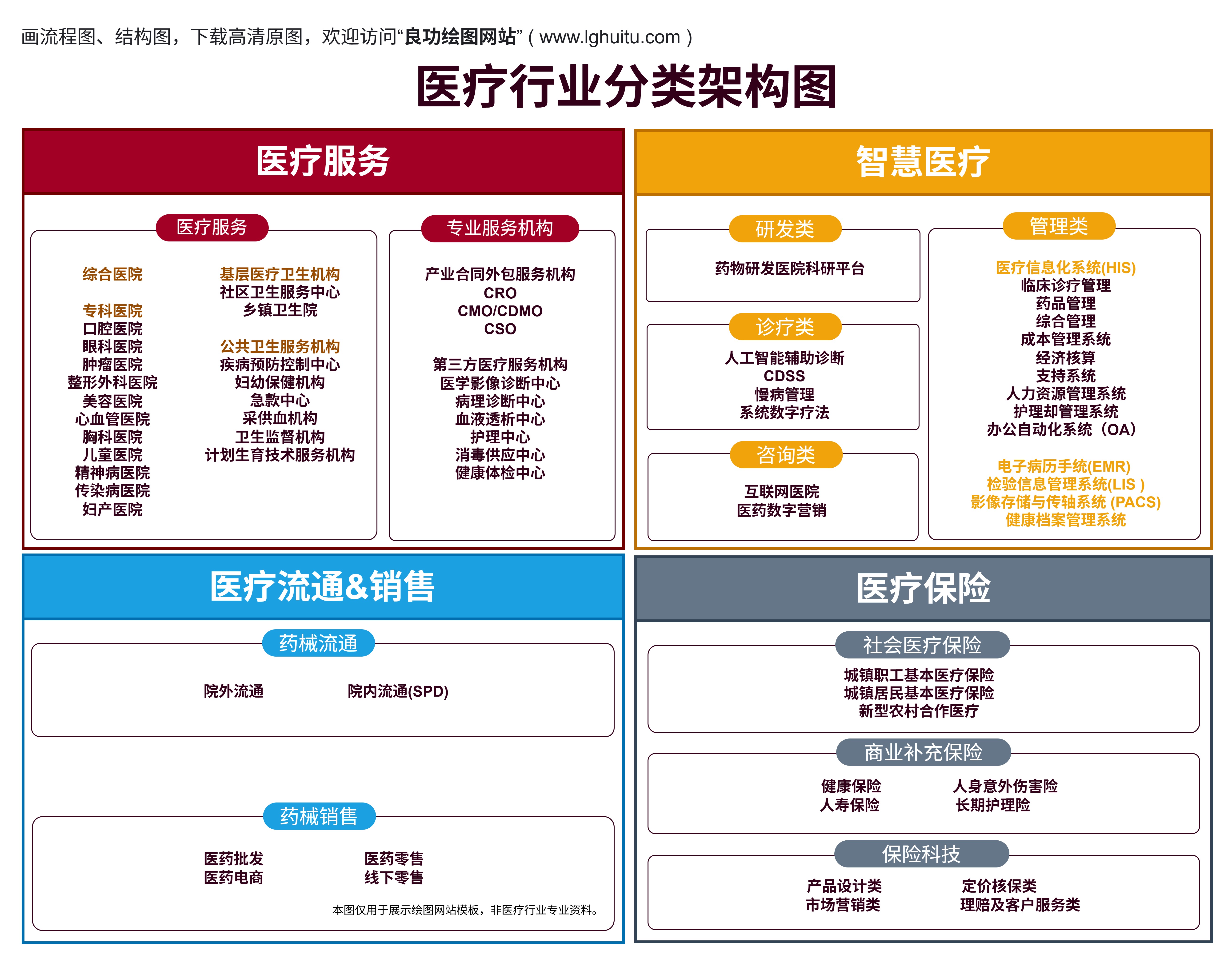
随着现代医学的飞速发展,医疗器械的种类繁多,涵盖了从基础的诊疗工具到高端的治疗设备。而根据我国医疗器械的管理规定,所有进入市场的医疗器械都必须经过注册审批。医疗器械的注册不仅是进入市场的法律要求,更是保障使用者安全、维护行业规范的重要环节。

医疗器械根据其风险程度的不同,分为一类、二类、三类。每一类的注册要求都有所不同,企业在进行注册前需要清楚了解每个类别的具体要求。
一类医疗器械是指那些风险较低、对人体安全性和有效性影响较小的医疗器械。典型的一类医疗器械包括一些简单的诊断工具、手术器械等。由于其风险相对较低,一类医疗器械的注册程序较为简单。
备案管理:对于一类医疗器械,通常不需要进行注册审批,而是通过备案管理的方式进行市场准入。企业只需在规定的时间内提交备案申请,并提供相关产品信息、质量管理体系等材料。
质量管理要求:一类医疗器械的生产企业需要建立和实施符合规定的质量管理体系,确保产品的生产符合相关的质量标准。
无临床试验要求:大部分一类医疗器械不需要进行临床试验。企业提供合格的生产记录和产品数据即可。
注册时间较短:备案通过的产品可以较快速地进入市场,一般无需等待长时间。
二类医疗器械是指那些风险较高、可能对人体安全性和有效性产生一定影响的器械。与一类器械相比,二类医疗器械需要更严格的监管和审核,确保产品的安全性和有效性。常见的二类医疗器械包括体外诊断试剂、医用超声设备等。
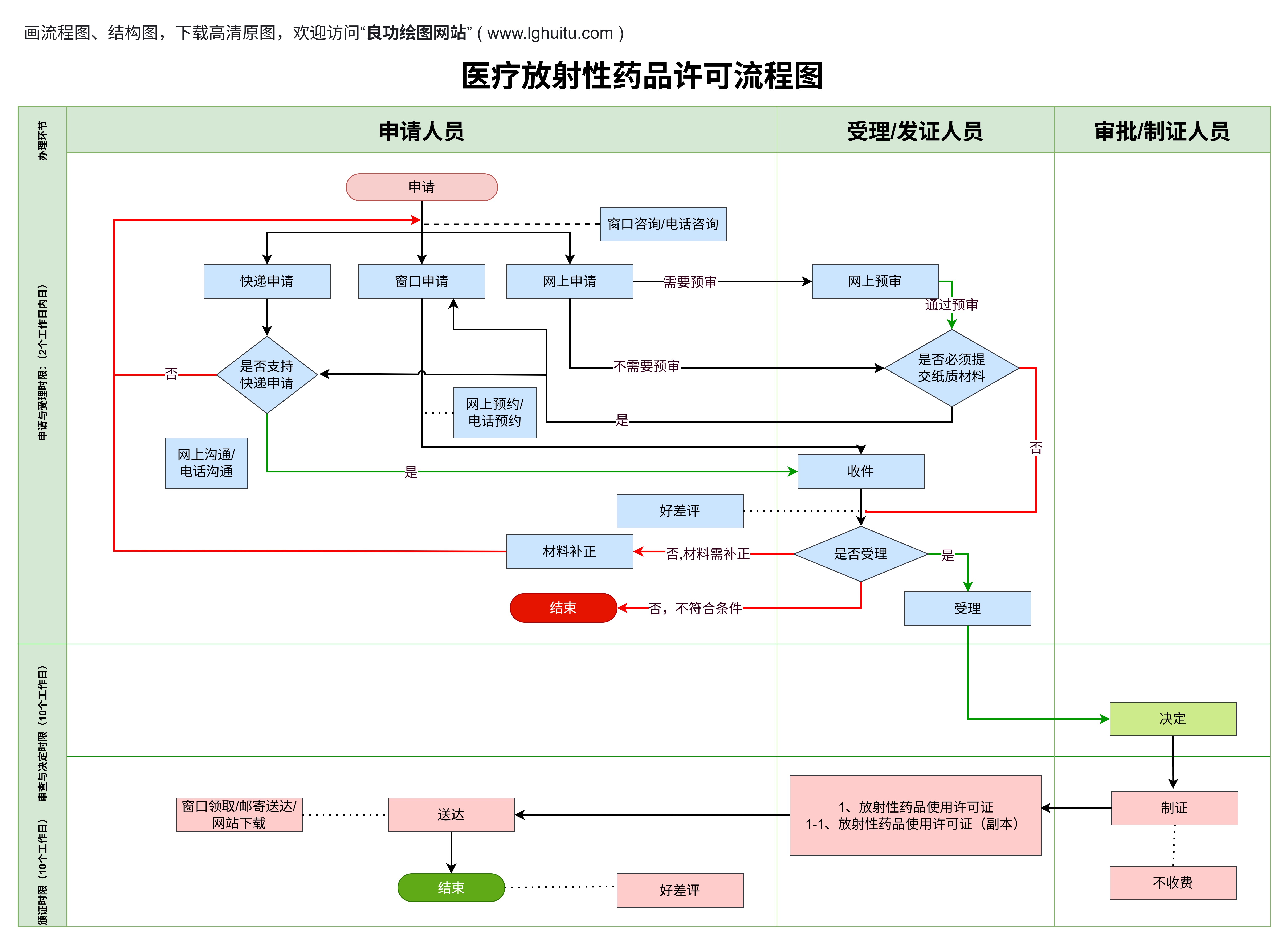
产品注册申请:企业需要向国家药品监督管理局(NMPA)提交注册申请,提供详细的产品资料,包括产品的技术参数、使用说明书、质量管理体系文件等。
临床试验要求:部分二类医疗器械需要进行临床试验,特别是那些用于治疗或诊断较为复杂疾病的器械。企业需要提交试验方案,并通过审批后进行临床试验。
风险管理:二类医疗器械企业需要进行详细的风险评估,并采取措施降低潜在风险。这不仅是注册要求,也是产品研发过程中必不可少的一步。
注册审核周期较长:二类医疗器械的审核过程较为复杂,时间通常较长。企业需提前做好相关准备,以免因审核周期影响上市时间。
三类医疗器械是指那些风险较高、可能对人体健康和生命安全产生重大影响的器械。这类器械通常是用于生命支持、重大疾病诊断和治疗的高端设备,如植入性医疗器械、生命支持设备等。由于其高风险性,三类医疗器械的注册要求是最为严格的。
严格的注册申请:三类医疗器械的注册申请流程是最为复杂的,企业需要提交大量的资料,包括详细的产品技术参数、临床试验数据、质量管理体系文件、生产工艺等。产品需要经过严格的审核,才能获得批准。
临床试验要求:几乎所有三类医疗器械都需要进行临床试验,并提交相关的临床试验报告。这些试验需要严格按照国际和国内的标准执行,以确保产品的安全性和有效性。临床试验的过程和数据必须经过相关部门的审核,方能继续进行注册。
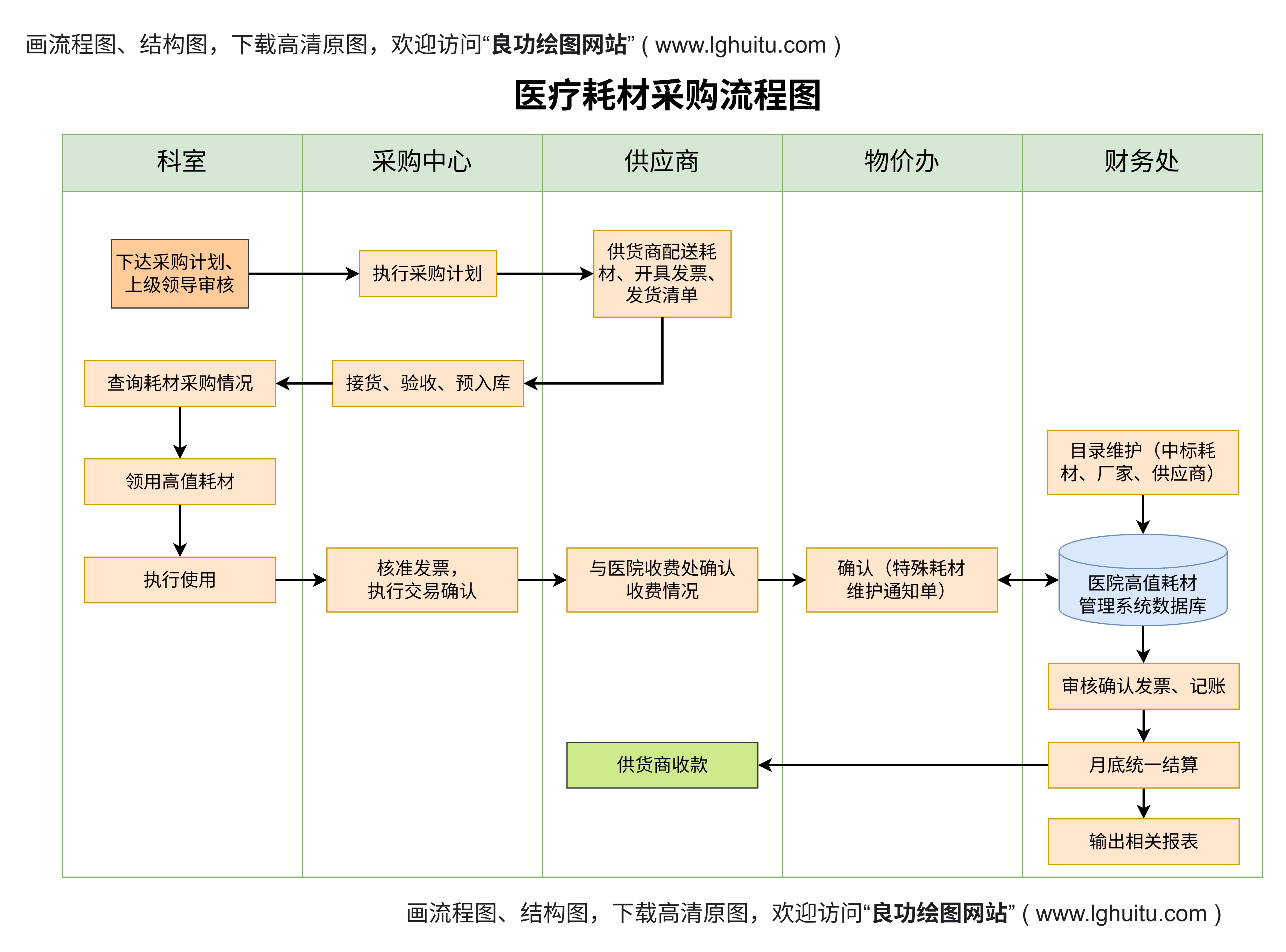
风险评估与控制:对于三类医疗器械,企业必须进行全面的风险评估,分析产品使用过程中可能存在的各类风险,并提出合理的风险控制方案。监管部门通常要求企业详细描述风险管理措施和应急处理预案。
注册周期长:由于三类医疗器械的复杂性和严格的审核要求,其注册周期通常较长,企业需要提前做好充分准备,确保各项资料的完整性和合规性。
风险等级:一类医疗器械的风险较低,二类和三类的风险逐渐增加,三类器械的风险最高,因此要求最严格。
审核程序:一类器械通过备案管理,二类和三类则需要提交详细的注册申请并接受严格的审查,尤其是三类器械,注册过程尤为复杂。
临床试验:只有二类和三类医疗器械需要进行临床试验,而一类器械大多无需进行。
注册周期:一类器械的注册周期相对较短,而二类和三类器械的审核周期则相对较长。
医疗器械的注册是一个复杂而严谨的过程,了解不同类别器械的注册要求,有助于企业在进入市场时更顺利、更高效地完成审批。无论是通过备案管理的一类器械,还是需要进行临床试验和风险评估的二类、三类器械,企业都需要提前做好规划,确保所有注册材料的完整性和合规性。希望本文为您提供了有价值的参考,帮助您顺利完成医疗器械注册,并进入广阔的市场。
