在中国,医疗器械的管理受到严格监管,尤其是二类医疗器械的审批流程。二类医疗器械被定义为“对人体有一定风险,但能通过合理的管理控制其风险的器械”。这类器械的种类繁多,涵盖了从诊断设备到治疗工具等多种产品,如血糖仪、心电图仪、超声波治疗仪等。想要将这些产品投放市场,首先需要通过国家药品监督管理局(NMPA)或其下属的地方监管机构的审批。二类医疗器械的申请流程究竟是怎样的呢?本文将为您详细解析。
二类医疗器械的申请并不是一个简单的过程,首先需要进行的是产品的初步准备工作。这包括以下几个方面:
产品分类确认:在申请之前,首先需要确定自己的产品属于哪一类医疗器械。二类医疗器械相对来说风险较低,但也需要遵循一定的监管要求。若产品分类错误,可能导致后续申请过程中出现问题,甚至遭到拒绝。
制定研发计划:企业需要明确研发产品的设计、生产和测试标准。二类医疗器械通常要求提供完整的技术资料和临床数据,证明产品在使用过程中安全有效。
ISO认证:许多二类医疗器械在申请时需要附带ISO认证,尤其是在生产过程中对质量控制有严格要求的产品。申请者需要确保产品在生产和质量管理方面符合国际标准。
申请二类医疗器械时,申请企业需要准备一系列的材料,确保资料齐全,符合国家法规。主要的申请材料包括:
产品说明书和技术资料:包括产品的设计理念、功能说明、适用范围、技术参数等,所有这些都需要详细列出,并符合监管要求。技术资料的准备是申请过程中最为复杂的环节之一,尤其是对于高精度的医疗器械产品。
质量管理体系文件:包括ISO13485认证,证明公司具有完善的质量管理体系,确保产品在生产、储存、运输等各个环节中都能保持高标准的质量控制。
临床试验数据:虽然部分二类医疗器械不要求进行临床试验,但某些产品在申请过程中依然需要提供临床数据,以证明其安全性和有效性。特别是一些有较大风险的医疗器械,临床数据至关重要。
其他辅助材料:如生产设备清单、企业营业执照、法人代表身份证明等。
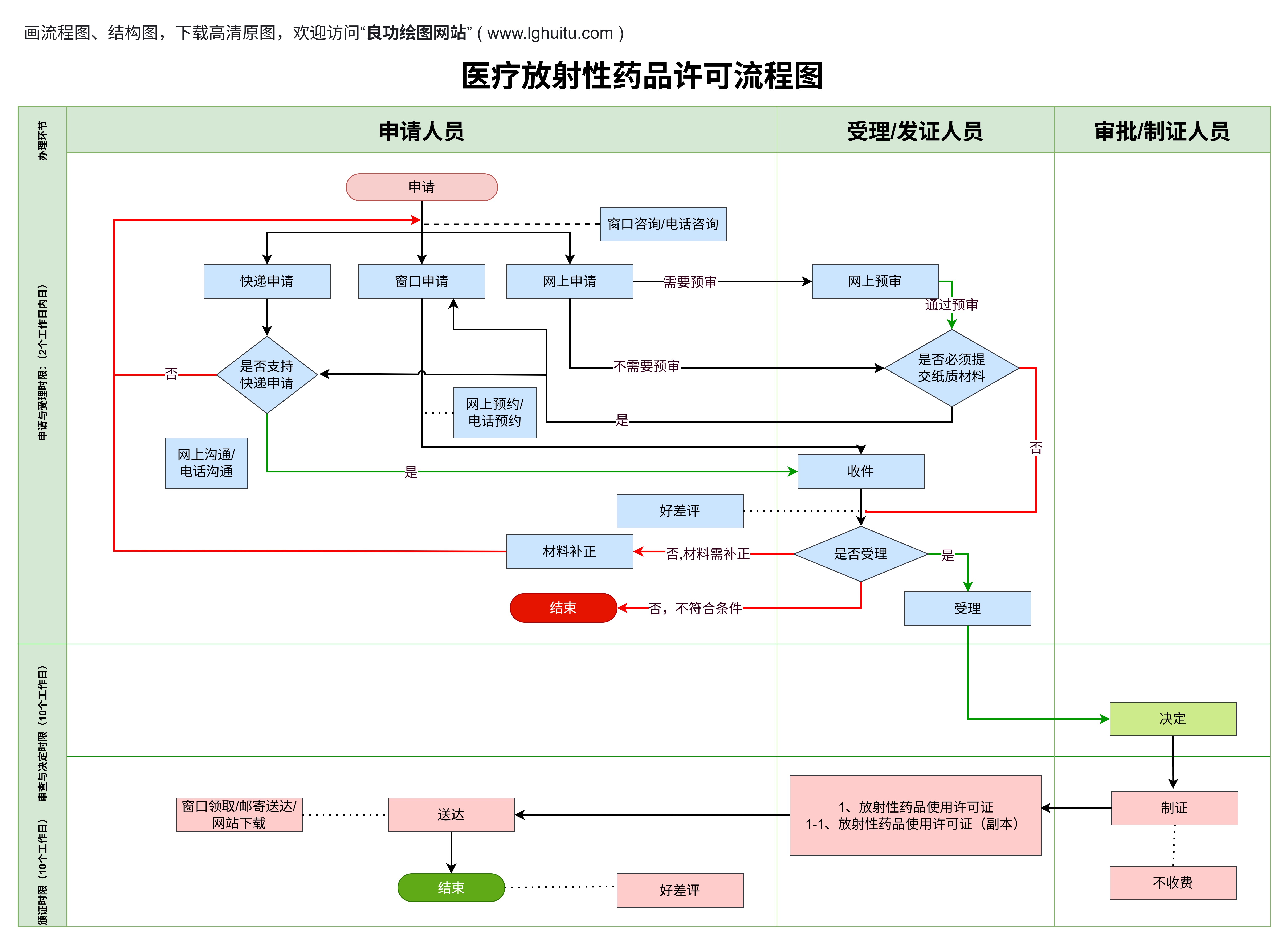
所有材料准备齐全后,企业就可以向当地药监局提交医疗器械注册申请。不同地区的监管机构可能在细节上有所不同,但总体的申请流程和材料要求是相似的。
线上申请:在中国,药品监督管理局推出了“医疗器械注册管理系统”,申请人可以通过该系统提交注册资料。在提交资料后,系统会对申请资料进行初步审查,如果资料完备,审核通过后,申请将进入下一阶段。
现场审核:对于一些涉及到生产设施、质量管理体系等的申请,药监部门可能会派员到企业进行现场审核,确认企业的生产条件、质量管理等是否符合相关规定。
申请材料提交后,药监局将进入详细审核阶段。通常,二类医疗器械的审核时间会相对较短,但具体时间会受到申请产品复杂度、监管局工作负荷等因素的影响。审核的内容包括以下几个方面:
材料审核:药监局会首先检查申请资料的完整性和规范性,确保所有文件、数据均符合相关标准。如存在问题,药监局将要求企业进行补充或修改。
技术评审:药监局会派遣专业评审人员对产品的技术资料、临床数据等进行详细审查,确保产品安全有效。如果产品涉及到较为复杂的技术问题,审核时间可能会更长。
现场检查:对于需要进行现场检查的企业,药监局会派人对生产条件、生产工艺、质量控制体系等进行实际检查,确保企业能够生产出符合标准的医疗器械产品。
经过严格的审核后,药监局会颁发医疗器械注册证书。注册证书是二类医疗器械在市场上合法销售和使用的凭证,标志着产品已通过国家的安全性与有效性审查。
产品上市前的准备:拿到注册证书后,企业还需要进行产品标签、说明书的规范设计,以符合药监局对于医疗器械产品包装的相关要求。每个产品的标签和说明书上都必须清晰标明使用方法、适应症、禁忌症、注意事项等关键信息,保障消费者的使用安全。
上市后的监管:二类医疗器械产品在上市后仍然需要接受监管部门的监督,确保其持续符合质量标准。这包括产品的定期抽检、生产过程的审查等。
二类医疗器械的申请流程虽然相对较为简单,但也需要企业具备一定的专业知识和经验。完整的产品技术文档、质量管理体系、临床数据等是审批过程中至关重要的环节。通过细心准备和严格遵守规定,企业不仅能够成功获得注册证书,更能够在激烈的市场竞争中脱颖而出。
