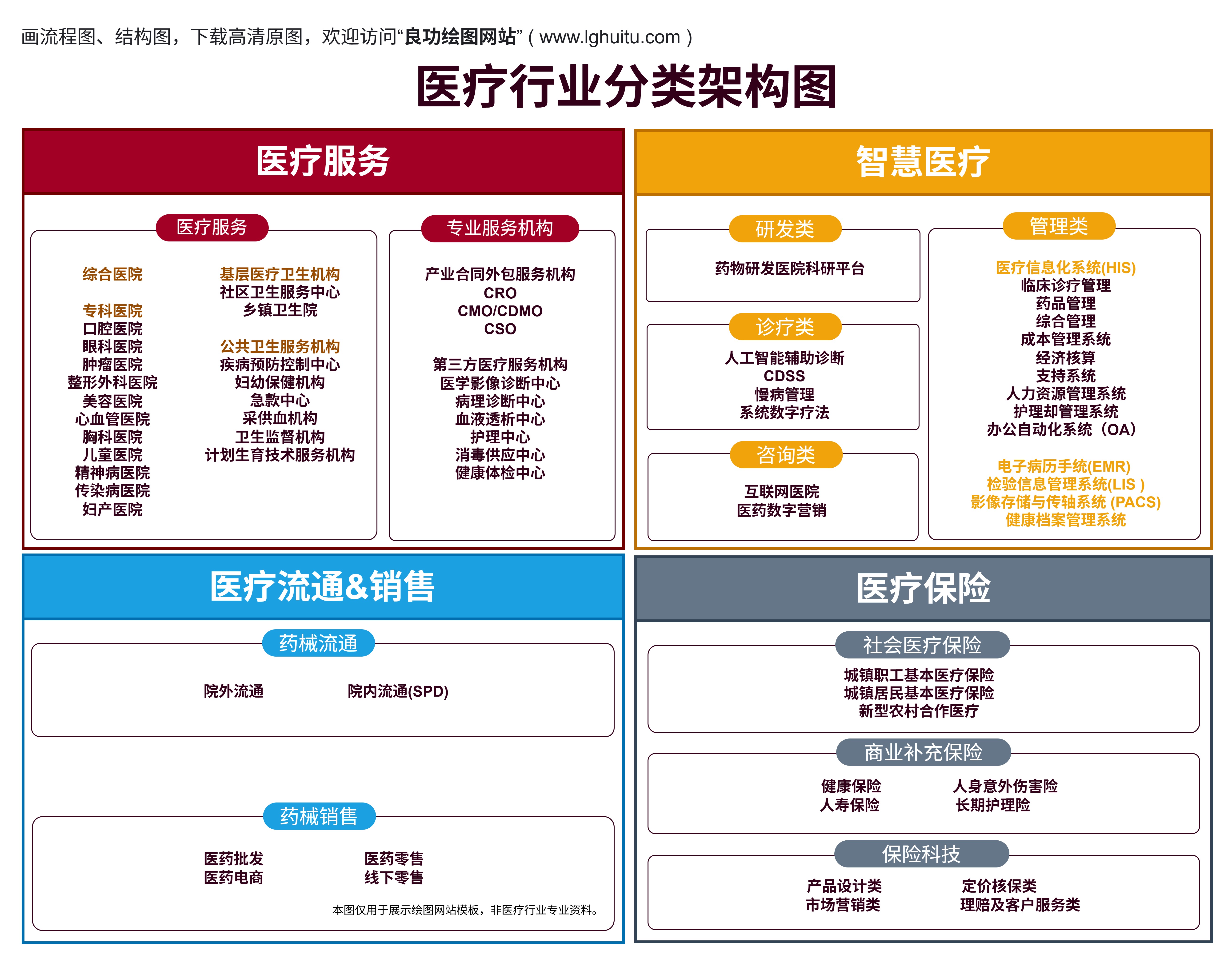
在中国,医疗器械行业的迅猛发展促进了现代医疗技术的进步,但同时也给医疗器械的安全性、有效性带来了严峻的挑战。为了确保每一款医疗器械能够真正服务于患者、提升治疗效果,国家对医疗器械的管理做出了严格要求。医疗器械的合法性与合规性,不仅关系到企业的生存与发展,还直接影响到患者的健康。

而在这其中,医疗器械的“三证”便成为了企业进入市场的“敲门砖”。这三证即是:医疗器械注册证、医疗器械生产许可证、医疗器械经营许可证。下面就详细解读这三证的含义、申请要求及其重要性。
医疗器械注册证是指国家食品药品监督管理局(NMPA,原CFDA)对医疗器械产品进行注册审批后,发放的证明该医疗器械符合国家标准并可以合法上市的证书。无论是国内生产还是进口医疗器械,只有获得注册证书,才能进入中国市场销售。
医疗器械注册证的获取是一个严格且系统化的过程。企业需要根据医疗器械的分类和风险等级,提交相关的技术资料、临床试验数据(如适用)以及产品说明书等。注册证的审批不仅仅是对产品质量的把关,还是对医疗器械可能带来的风险进行全方位评估的过程。
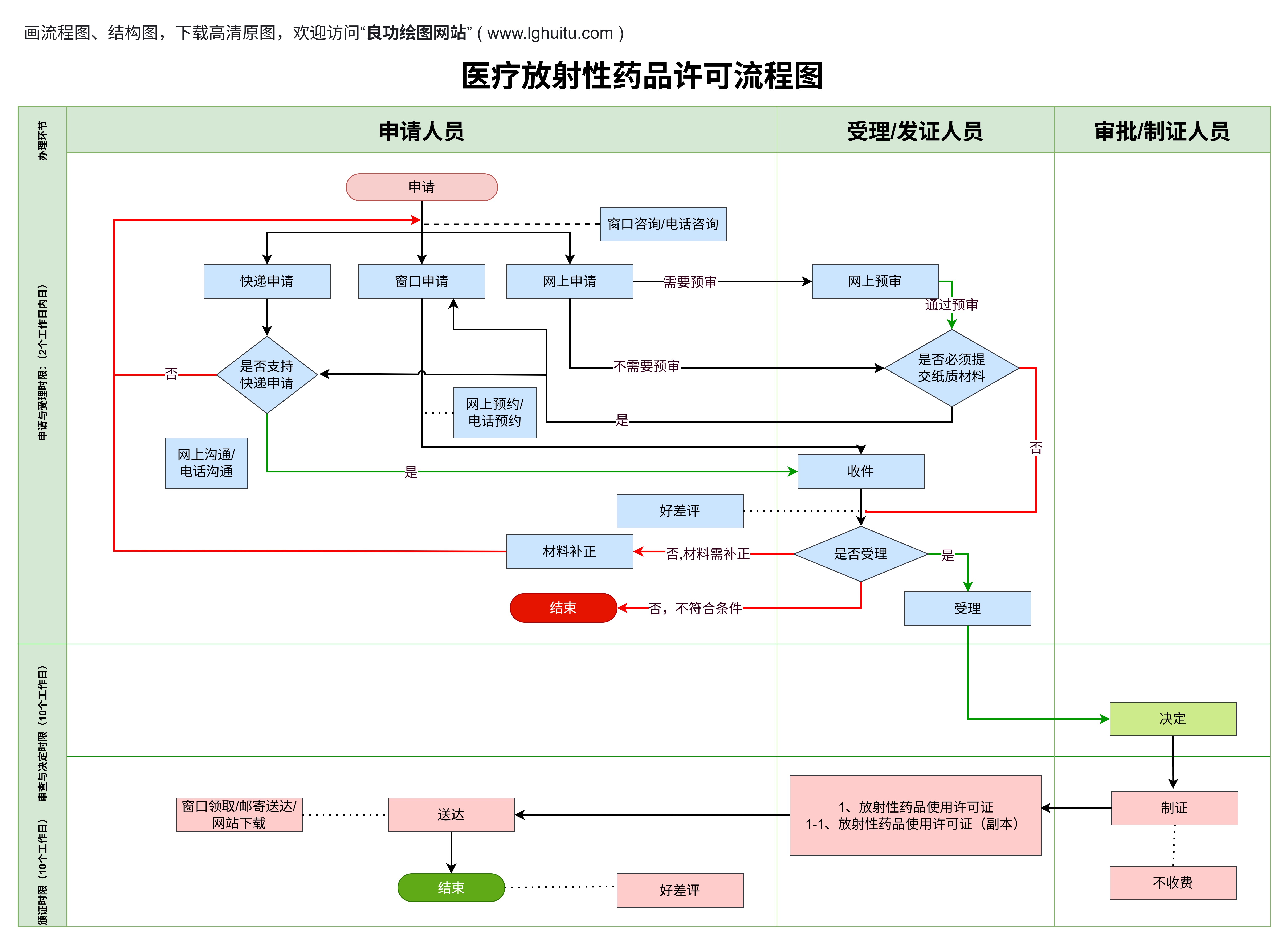
具体来说,注册证的获取不仅能够证明产品的合规性,还能为产品提供市场准入的保障。对医疗器械生产企业来说,拥有注册证代表着其产品能够合法销售,且其生产的每一件医疗器械都需要符合注册证中的规定。
与注册证不同,医疗器械生产许可证更多地关注的是生产过程的合规性。根据《医疗器械监督管理条例》规定,所有医疗器械的生产单位必须依法申请并获得医疗器械生产许可证,才能进行生产、加工与组装。
医疗器械生产许可证的审批,主要是对生产环境、生产设备、技术人员、质量管理体系等方面进行严格的审查,确保企业具备生产符合国家标准医疗器械的能力。对于企业来说,获得生产许可证意味着其具备了生产合法医疗器械的资质,并且其生产过程符合质量控制和安全规范。
生产许可证的申请过程涉及多个环节,包括工厂现场检查、生产工艺评估、质量管理体系审核等。经过严格的审查,生产许可证的颁发确保了企业在生产医疗器械时,能够遵循标准化的管理流程,有效防止生产过程中出现不合格产品。

除了注册和生产,医疗器械的销售环节也同样需要合法合规。医疗器械经营许可证,顾名思义,是指从事医疗器械销售、批发、零售等业务的单位,必须向相关监管部门申请并获得的许可文件。无论是线上销售还是线下销售,只有具备医疗器械经营许可证的企业,才能合法地从事医疗器械的流通业务。
经营许可证的申请同样需要经过严格的审核程序,涉及到的审查内容包括:企业的经营场所、销售人员的资质、产品的贮存条件、物流管理等。经营许可证的作用不仅是确保销售单位能够合法经营,更是保障消费者权益的重要环节。只有持有经营许可证的企业,才能合法地进行产品销售,确保消费者购买到符合标准的医疗器械。
医疗器械的“三证”制度,实际上是从三个环节来保障医疗器械的合法性和安全性。从产品的合规性到生产过程的规范性,再到销售环节的合规性,每一项证书都是保证医疗器械在市场中流通时能够符合法律法规的要求。
这三证各自独立,却又紧密联系。例如,若没有医疗器械注册证,生产和销售环节将无从谈起;没有生产许可证,即便有注册证,也无法实现生产;没有经营许可证,即使产品已经生产完成,也无法顺利进入市场。因此,医疗器械三证之间相辅相成,共同确保了医疗器械的安全性、有效性与合规性。
在中国,医疗器械行业的监管越来越严格。企业在进入市场之前,必须首先通过“三证”认证,才能确保产品顺利投放市场。对于医疗器械企业而言,获得这三证不仅是合规的必要条件,更是提升品牌信誉、开拓市场的强有力保障。
医疗器械行业充满机遇,也充满挑战。想要在这片广阔的市场中立足,除了技术创新与产品质量的保证外,合规性同样不可忽视。通过完善的“三证”制度,企业不仅能提高市场竞争力,还能为患者的健康保驾护航。因此,医疗器械企业要注重三证的办理和维护,把合规管理作为企业发展的基础,才能在激烈的市场竞争中占得先机。