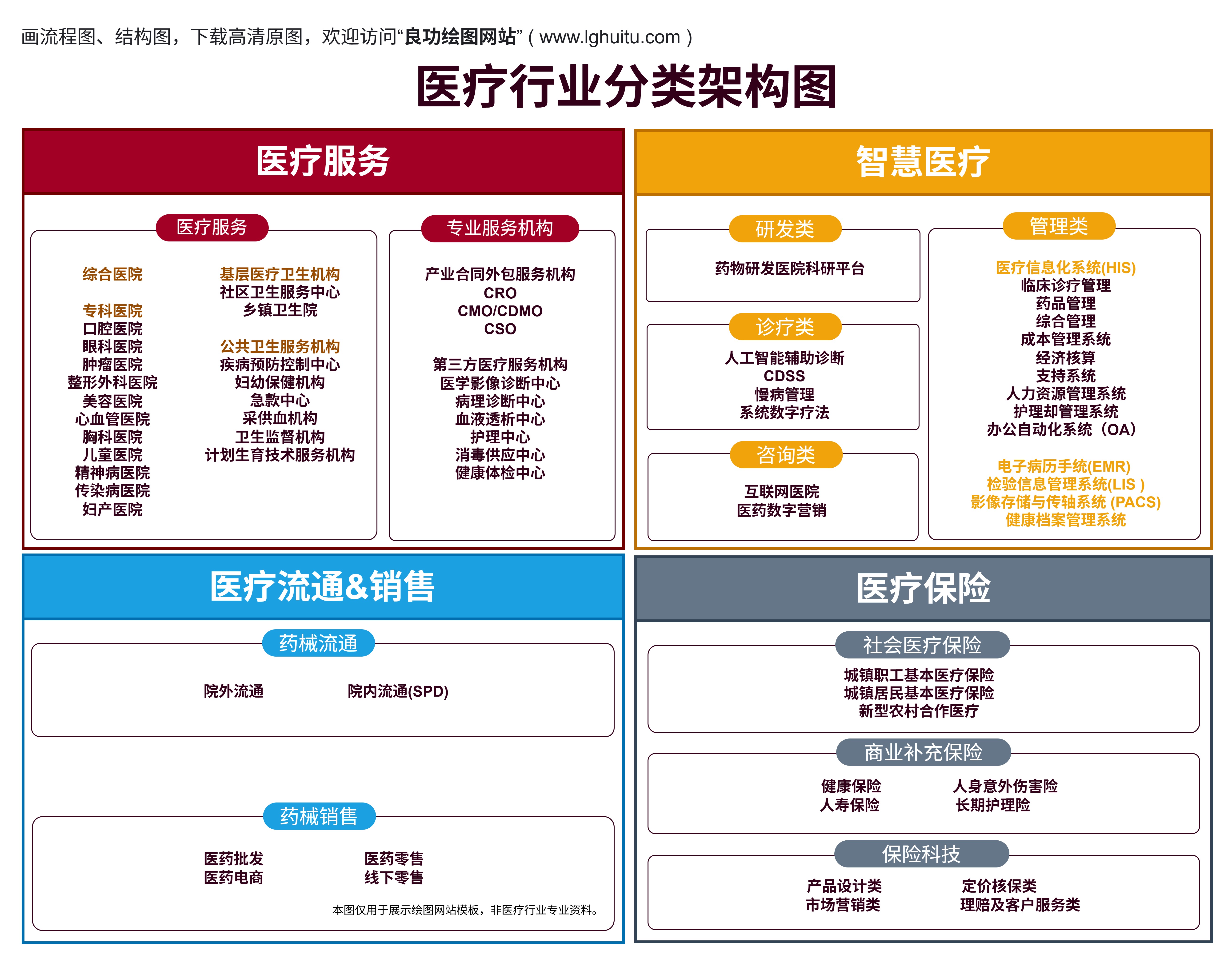
随着我国医疗健康产业的不断发展,医疗器械在日常生活中的应用越来越广泛。医疗器械的市场准入并非一蹴而就,企业在推广和销售医疗器械前,必须完成一项重要的任务——医疗器械注册。医疗器械注册不仅是进入市场的前提,也是确保产品质量、安全、有效的重要环节。医疗器械注册到底是怎样的一个过程呢?本文将为您揭秘医疗器械注册的全流程,帮助企业顺利走上合规的道路。

医疗器械的种类繁多,从简单的手术器械到复杂的影像设备,每一类产品的注册要求都不同。为什么注册如此重要呢?医疗器械作为直接影响到人类健康的产品,必须经过严格的审批程序。只有通过国家药监局(NMPA)认证的产品,才能保证其安全性和有效性,避免市场上出现不合格的医疗器械,保障公众的健康。

医疗器械注册是企业进入市场的法定程序。如果没有注册,企业将无法获得销售许可,产品也无法进入医院或药店。因此,注册过程的顺利与否,直接影响到企业的市场布局和商业前景。
根据我国的《医疗器械监督管理条例》,医疗器械分为三类:一类、二类和三类。不同类别的医疗器械在注册要求和审批程序上有所不同。
一类医疗器械:此类器械风险较低,通常是简单的诊断或治疗工具,如血糖仪、温度计等。对于一类器械,企业需要向当地药监部门提交注册申请,并提供基础的产品信息、生产工艺等文件,经审批后即可上市。
二类医疗器械:此类器械的风险适中,涉及一些较为复杂的诊疗设备,如超声波设备、常规体外诊断试剂等。二类器械的注册需要进行临床试验,提供相关的技术资料和临床数据,且需要经过药监局审批。
三类医疗器械:三类器械属于高风险产品,通常用于生命支持、诊断精度较高的医疗器械,如心脏起搏器、影像诊断设备等。对于三类器械,注册过程较为复杂,除了提供详细的技术资料和临床试验数据外,还需要进行风险评估和认证审查。企业需要提交完整的技术文档、生产流程以及临床验证资料,并且可能需要接受药监部门的现场检查。
不论是哪一类医疗器械,注册的基本流程通常包括以下几个主要步骤:
产品分类与注册准备:在进行医疗器械注册前,企业首先需要明确产品的类别,根据产品的风险等级选择合适的注册路径。企业还需准备相关的技术资料,包括产品说明书、生产工艺、质量控制等方面的资料。
注册申请提交:一旦准备好相关资料,企业需要向国家药监局或地方药监部门提交注册申请。此时,申请文件的完整性和准确性至关重要,任何资料的遗漏或不准确都会导致注册的延误或被拒。
技术审查与验证:药监局收到注册申请后,会对提交的技术资料进行审查。对于二类和三类医疗器械,药监局可能还会要求企业提供临床试验报告,并对产品的安全性和有效性进行评估。药监局还可能要求企业对产品进行现场检查。
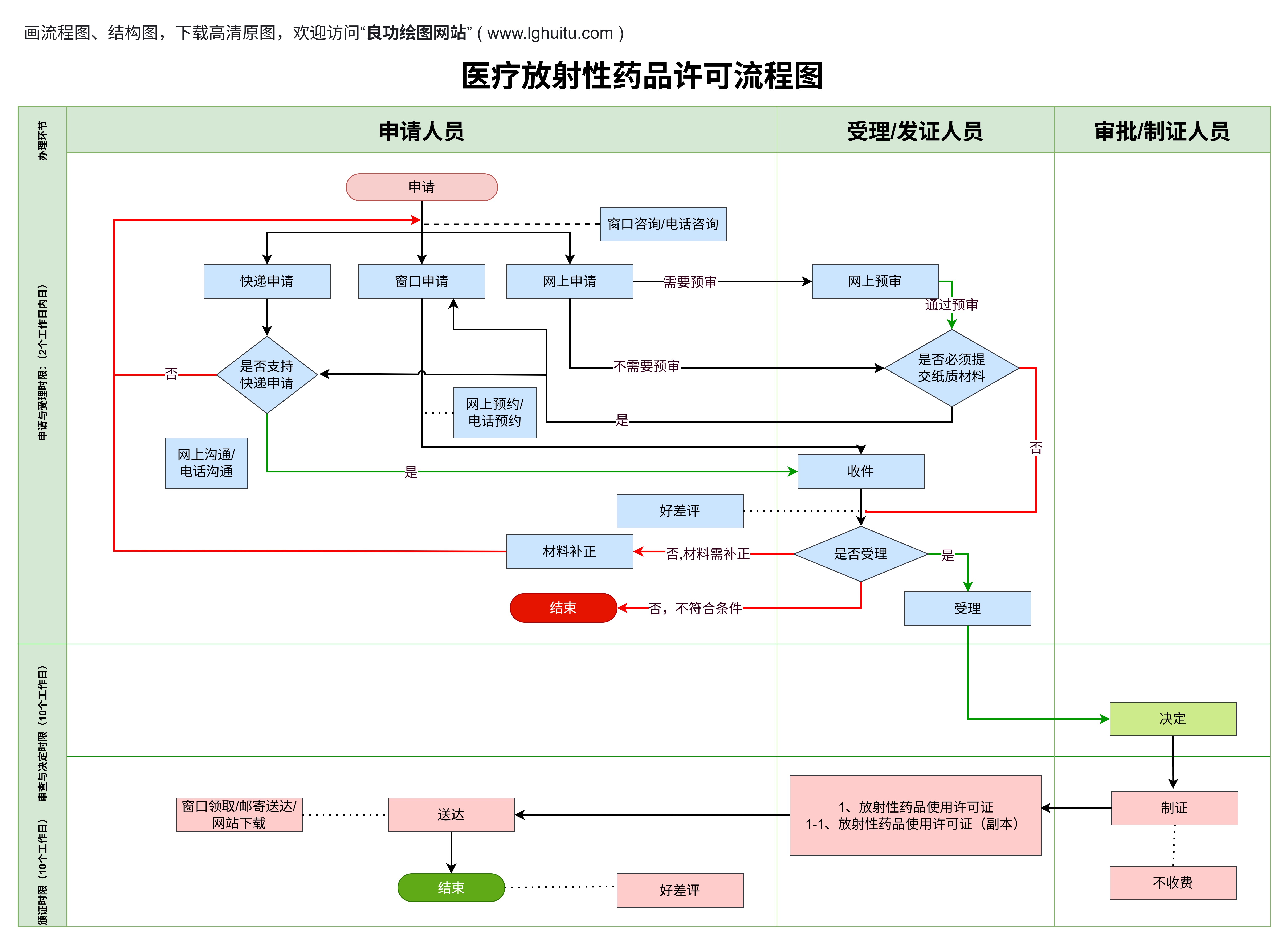
注册证书颁发:当所有资料审查通过后,药监局将颁发《医疗器械注册证》,此证书是产品合法销售的证明。获得注册证书后,企业即可将产品推向市场,开始生产和销售。
资料准备的全面性:注册申请时,所有的技术文档和资料必须真实、准确、完整。尤其是对于二类和三类器械,临床试验报告、产品性能测试报告等都必须符合相关标准,否则可能会导致注册延误或拒绝。
临床试验的重要性:对于二类和三类医疗器械,临床试验是注册过程中不可或缺的一环。企业需选择符合国家标准的医疗机构进行试验,确保试验数据真实可靠。临床试验不仅有助于证明产品的安全性和有效性,还能为产品的市场推广打下坚实的基础。
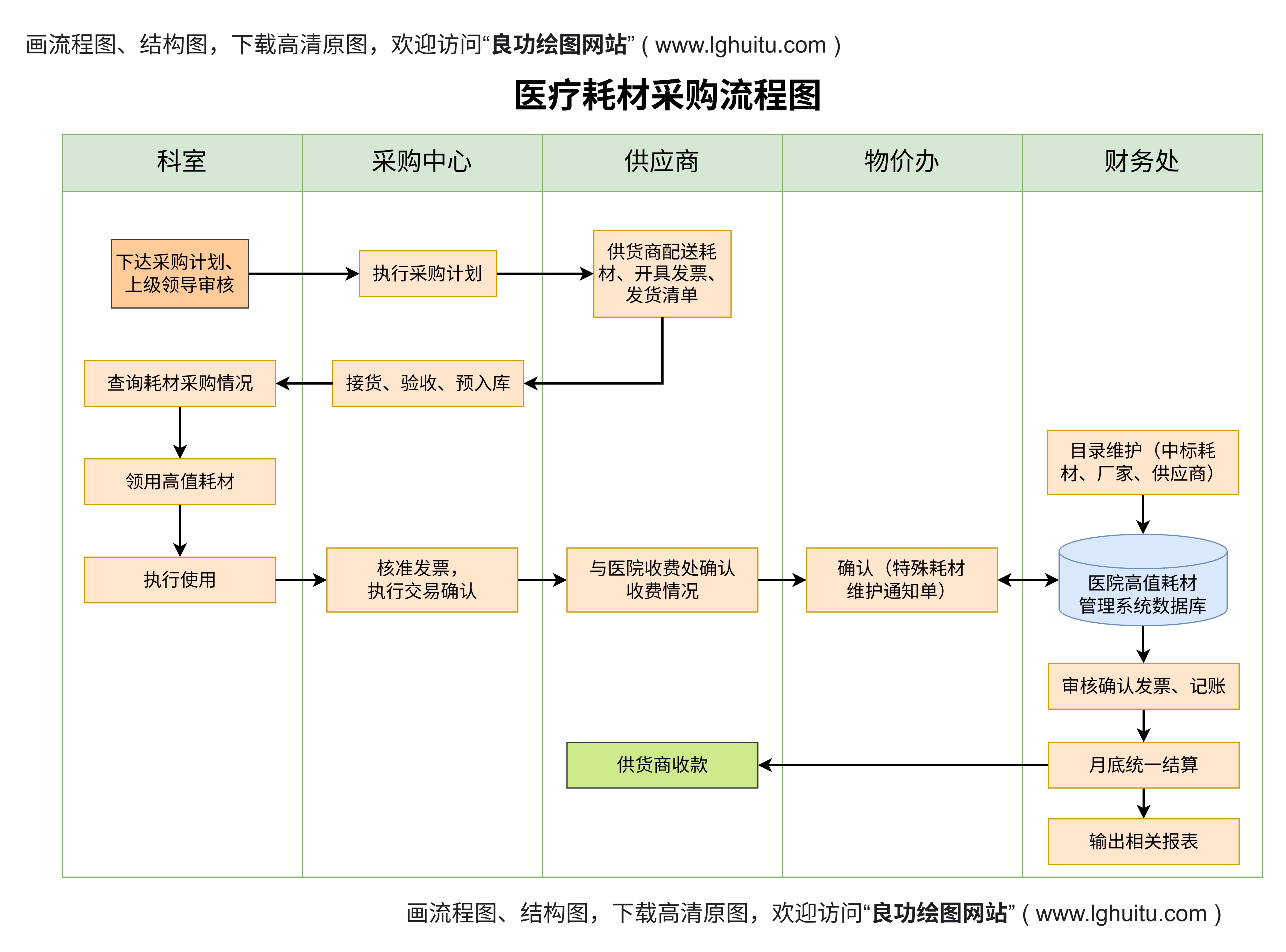
药监局的现场检查:在注册过程中,药监局可能会对企业的生产场地、设备设施进行现场检查。企业需要提前做好生产环境的整改和设备的验证,确保符合药监局的要求。
注册后的持续合规:获得注册证书后,企业不仅需要按照批准的标准生产产品,还需要进行持续的质量控制和监督。药监局对已注册医疗器械的后续监管也很严格,企业应定期进行产品质量检测和报告,确保产品始终符合相关的法规要求。
医疗器械注册是一个复杂且严格的过程,但通过科学的规划和准备,企业可以在此过程中减少不必要的时间浪费,尽早获得注册证书。以下是几个加速注册流程的小贴士:
提前与药监局沟通:在开始注册之前,企业可以通过咨询药监局,了解最新的政策动态和注册要求,避免因政策变化而影响注册进度。
选择专业的注册代理机构:医疗器械注册涉及大量的法规和技术要求,对于许多企业而言,可能缺乏足够的经验和专业知识。选择一家具有丰富经验的注册代理机构,可以帮助企业提高注册效率,降低注册风险。
做好产品的质量控制:从研发到生产,企业应注重产品质量的控制,确保每个环节符合标准。这不仅有助于顺利通过注册审查,还能提高产品的市场竞争力。
通过以上步骤和注意事项,企业可以有效地推动医疗器械注册的进程,为产品的顺利上市打下坚实的基础。无论是在产品研发、临床试验,还是在资料提交和现场检查中,合理规划、周密准备和持续合规将是企业成功的关键。