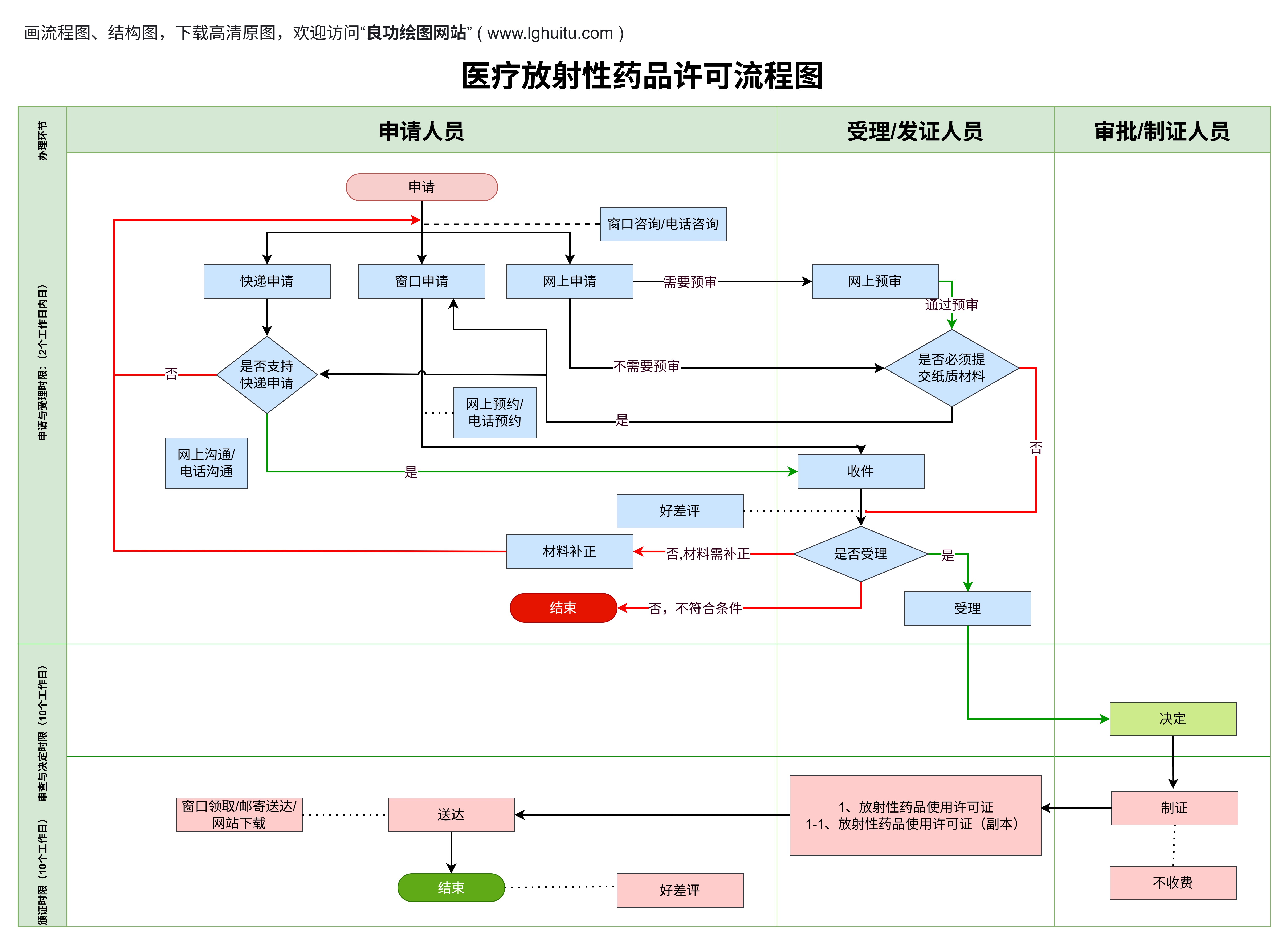
在如今医疗行业日益发展的背景下,医疗器械的市场需求不断攀升。对于医疗器械生产企业来说,获取医疗器械生产许可证是迈向合法合规经营的关键一步。无论是初创公司,还是已有一定规模的企业,办理医疗器械生产许可证都需要经过一系列严格的流程。如何才能顺利完成这个过程呢?

我们需要了解什么是医疗器械生产许可证。医疗器械生产许可证是由国家药品监督管理局(NMPA)发放的一种资质证明,旨在确保医疗器械的生产企业符合相关法规和标准。获得此证书后,企业方可在合法范围内开展生产、销售、经营等相关业务。可以说,医疗器械生产许可证是企业进入医疗器械行业、取得市场竞争力的基础。

在正式申请医疗器械生产许可证之前,企业需要完成以下几个准备工作:
包括有固定的生产场地、符合生产要求的设施和设备、具备符合国家标准的质量管理体系,以及具有一定规模和稳定经营的能力等。生产场地和设备应符合《医疗器械生产质量管理规范》(GMP)等相关规定。
根据《医疗器械生产质量管理规范》,企业必须建立健全质量管理体系,并设立专门的质量管理部门。企业要进行体系文件的编写与实施,确保从研发到生产的全过程都能够得到有效管理和控制。
企业需要聘用符合资格要求的技术人员,包括注册质量负责人、注册生产负责人等关键岗位。这些人员需要具备相关的技术知识和资质。
企业在确保自身符合基本条件后,下一步便是向当地的药品监督管理局提交申请材料。常见的材料包括:
企业的法人营业执照复印件需要提交,并确保其经营范围涵盖了医疗器械的生产或相关业务。
企业应提交完整的质量管理体系文件,包括质量手册、操作规程等,以证明企业具备完善的质量管理体系。
提交关于生产场地和生产设备的相关证明材料,确保其符合生产条件。
提供相关技术人员的资质证明,包括生产负责人和质量负责人等。
在申请过程中,企业还需提交具体产品的技术资料和说明书,以便监管部门评估产品的技术性能和合规性。
当所有材料提交完毕后,药品监督管理局将安排对企业生产现场进行核查。核查的主要内容包括:
检查企业生产环境是否符合GMP等相关规范,确保生产条件适宜。
核查企业生产线、设备及设施是否符合标准要求,是否具备生产所需的基本条件。
监管部门将审查企业是否按照提交的质量管理体系文件进行生产,确保各项管理措施得以落实。
如果在核查过程中发现问题,监管部门将要求企业整改并进行复查。因此,企业在申请前一定要做好充足的准备,避免不必要的麻烦。
完成现场核查后,药品监督管理局将对企业提交的材料和现场核查情况进行审核。审核的结果将决定是否批准企业的医疗器械生产许可证申请。如果企业符合所有的法规要求,监管部门将颁发医疗器械生产许可证。
虽然企业成功获得了医疗器械生产许可证,但这并不意味着可以高枕无忧。为了确保产品质量的持续稳定,企业还需进行以下几方面的管理:
医疗器械生产企业需要定期进行内部质量管理的审查与改进,不断提升生产工艺和质量控制能力,以适应法规和市场的变化。
药品监督管理局将定期对企业进行监管检查,以确保其生产活动符合相关法规要求。企业必须积极配合检查,整改发现的问题,确保不违反法律规定。
企业如果发生生产设备、人员或产品变更等情况,必须及时向监管部门报告,并提交相关的变更或补充材料。否则,企业的生产许可证可能会被暂停或撤销。
医疗器械生产许可证并非永久有效,通常有一定的有效期。企业需要在许可证到期前进行续期申请,以确保合法的生产资质不受影响。在续期申请过程中,企业需提交相关的经营状况和质量管理报告,证明其在生产过程中始终保持合规。
在办理医疗器械生产许可证的过程中,企业需要特别注意以下几点:
确保企业具备高水平的质量管理能力,符合《医疗器械生产质量管理规范》的要求,以便顺利通过现场核查。
企业应根据药品监督管理局的要求,提前准备好所有申请材料,并确保资料齐全、准确无误,以避免因材料不全而影响审批进度。
医疗器械是直接关系到患者健康和生命安全的产品,企业必须严格遵守相关法规和标准,确保生产过程中的每一环节都符合国家的要求。
医疗器械生产许可证的办理是一个复杂而繁琐的过程,但它却是企业合法经营的基础和保障。通过详细的准备工作、规范的质量管理体系和严谨的申请流程,企业不仅能顺利获得许可证,还能在激烈的市场竞争中脱颖而出。希望每一个有意从事医疗器械生产的企业都能按照规范要求,顺利取得医疗器械生产许可证,早日实现市场突破。