在医疗器械领域,产品根据其风险等级的不同被分为一类、二类和三类。每一类产品都需要申请相应的医疗器械许可证,且每类产品的管理要求和审批程序有所不同。了解这些分类及其差别,对于医疗器械生产企业来说至关重要。今天我们就来探讨医疗器械一类、二类和三类产品许可证的具体差异。

一类医疗器械是指风险较低的医疗产品。根据《医疗器械监督管理条例》的规定,一类医疗器械是指对人体安全性和有效性不产生重大风险的产品。由于其风险较低,监管要求相对宽松,企业在生产和销售这些产品时,所需的审批手续也相对简单。

对于一类医疗器械,企业只需进行产品备案,并不需要进行复杂的临床试验或者严格的质量控制要求。这也意味着一类医疗器械的市场准入门槛较低,适合小型企业和初创企业进入。这并不意味着企业可以忽视质量管理,仍需符合基本的生产和质量管理标准。
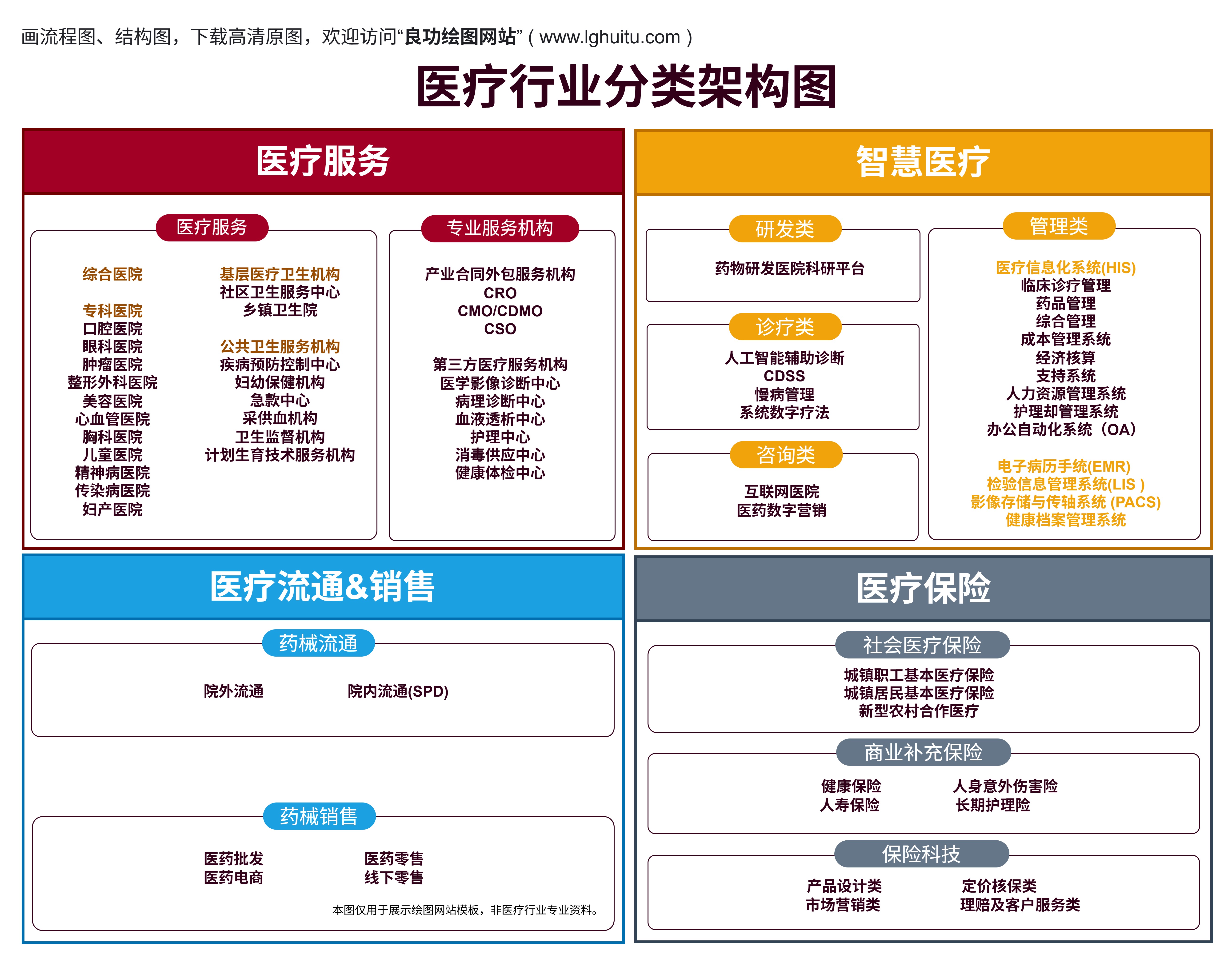
常见的一类医疗器械包括:医用绷带、一次性注射器、体温计、眼科用镜片等。这些产品通常不会对人体健康造成重大威胁,因此其监管要求也相对简单。
与一类产品相比,二类医疗器械的风险相对较高。二类医疗器械是指用于人体,可能对人体安全性和有效性产生一定风险,但通过有效的控制措施可以确保其安全性和有效性。为了确保这类器械的使用安全,相关部门对二类医疗器械的管理较为严格。
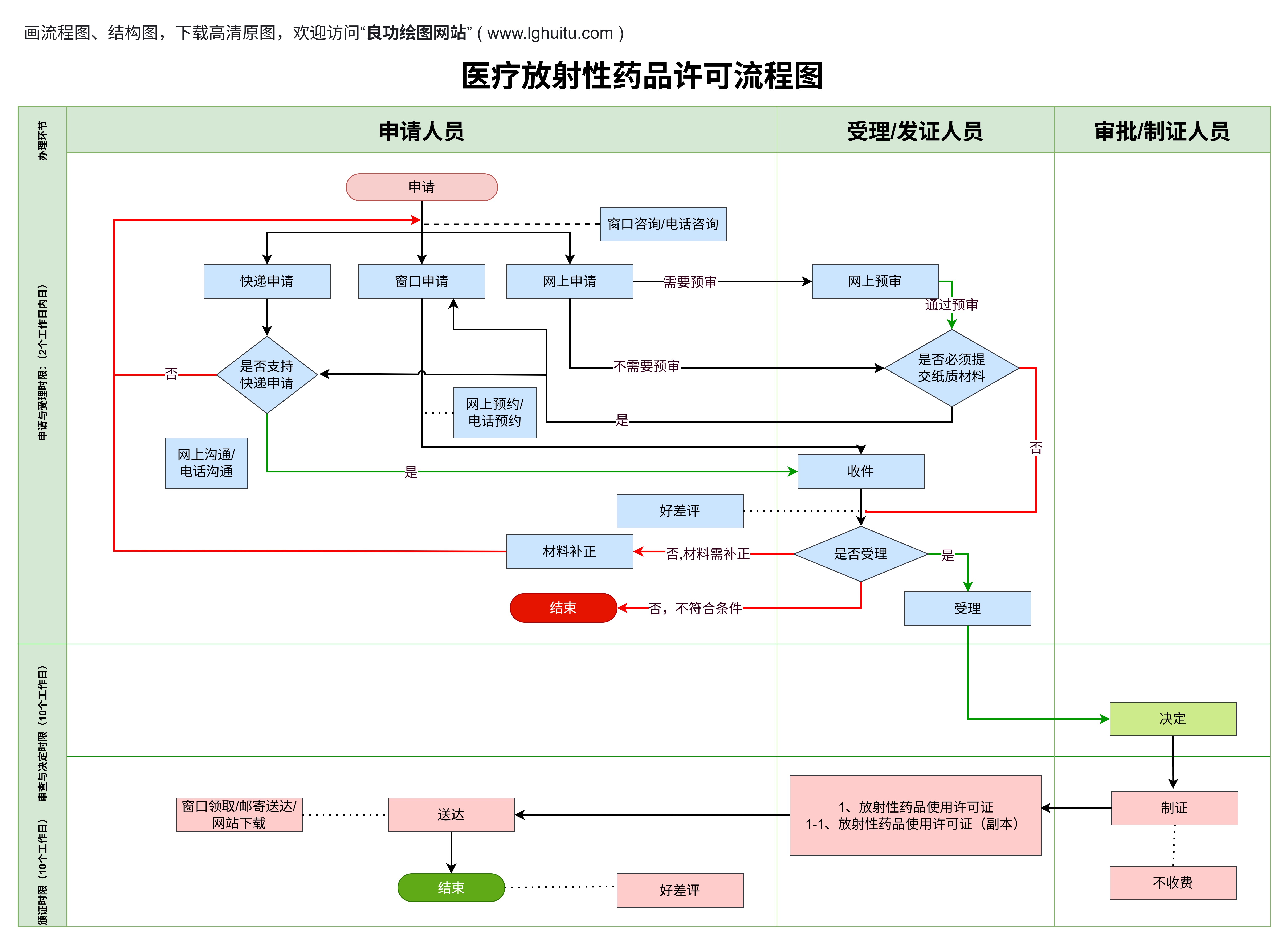
对于二类医疗器械,企业除了需要备案外,还需进行注册,并提供更加详细的产品资料,包括临床试验数据、技术文献、质量控制标准等。注册过程较为繁琐,审核时间较长,但这也是为了确保产品的安全性与有效性。
二类医疗器械的管理要求更加严格,企业需要建立更加完善的质量管理体系,并进行严格的产品检验和检测,以确保每一批产品都符合安全标准。常见的二类医疗器械包括:输液泵、CT扫描仪、血糖仪等。
从上述分析可以看出,一类和二类医疗器械的主要区别在于产品的风险等级和监管要求。一类医疗器械由于风险较低,审批流程较为简单,企业备案即可。而二类医疗器械则需要进行更为严格的注册,且审批过程更加繁琐,企业必须提供更多的技术资料和安全性数据。二类产品的市场准入门槛较高,企业需要具备较强的研发和生产能力。
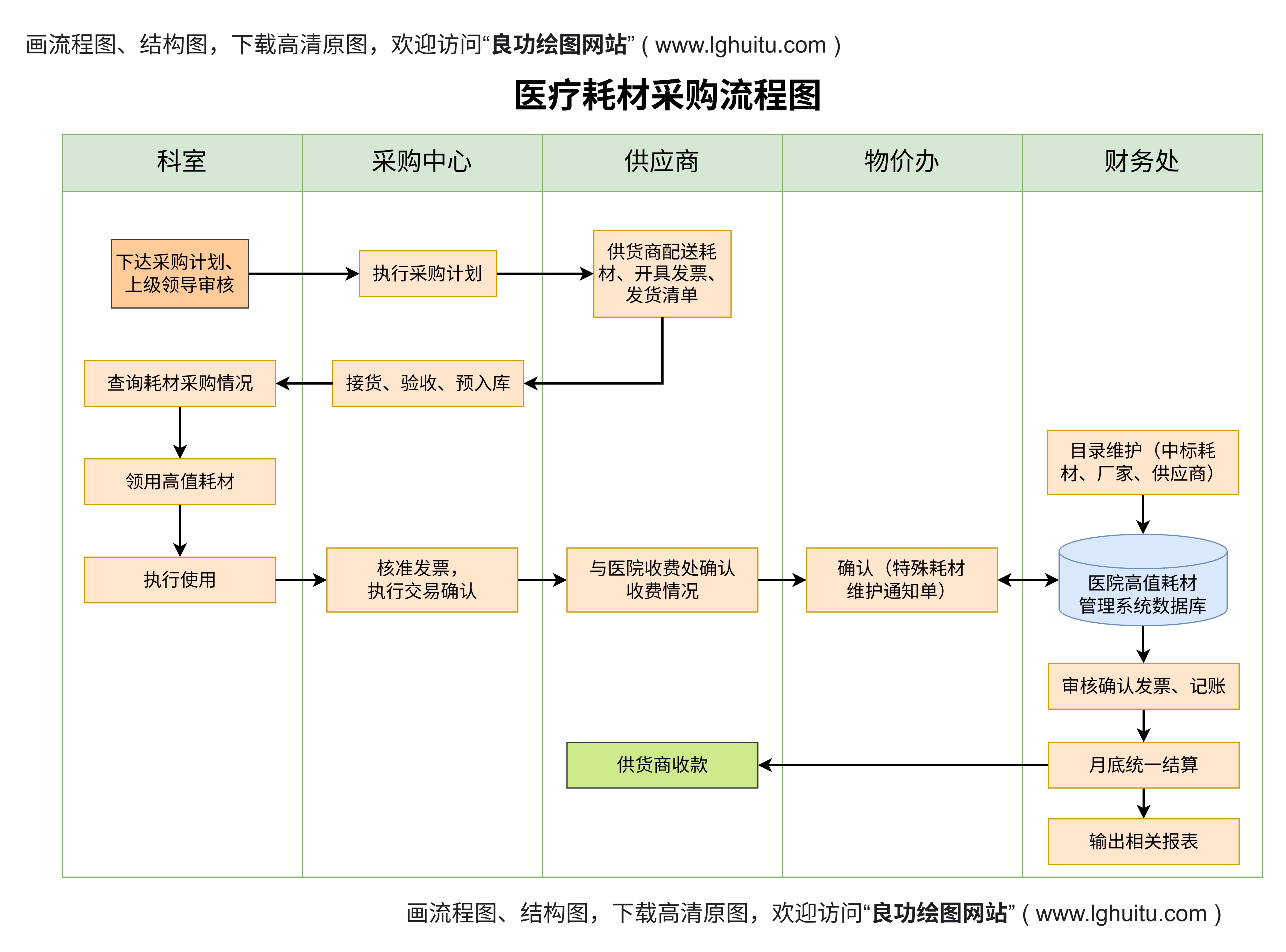
三类医疗器械是指对人体安全性和有效性具有较大风险的医疗器械。这类器械如果出现问题,可能对人体造成严重伤害或危害,因此对其进行严格的监管和控制是十分必要的。三类医疗器械通常涉及到生命支持、重大疾病治疗等领域,因此其审批程序和市场准入要求是最为严格的。
与一类、二类医疗器械不同,三类医疗器械需要进行临床试验和详细的安全性评估。企业在申请三类医疗器械许可证时,必须提供临床试验数据、产品的长期稳定性研究数据以及详细的质量管理体系文件。这些资料将帮助监管部门全面评估产品的安全性和有效性,以确保其对患者的安全性。
三类医疗器械的审批流程相对复杂,通常需要较长的时间,而且审批费用也较高。因此,申请三类医疗器械许可证对于企业来说是一个重大的挑战,要求企业在技术、资金、研发能力等方面具备较强的实力。
从管理的角度来看,一类、二类和三类医疗器械的最大区别在于风险等级和监管的严苛程度。三类医疗器械由于其潜在风险较大,因此需要进行详细的临床试验和严格的安全性评估,其审批过程也是最为严格的。二类医疗器械虽然风险较高,但并不需要进行全面的临床试验,只需提供相关的技术数据和产品资料。相比之下,一类医疗器械的审批过程相对简单,企业只需要进行备案即可。
随着产品的分类提升,企业在质量管理方面的要求也逐步增加。从一类到三类,企业需要建立更加完善的质量管理体系,并且严格遵循GMP(良好生产规范)等标准,确保每一件医疗器械产品的安全性和有效性。
医疗器械许可证一类、二类和三类的差别,主要体现在产品的风险等级以及相应的监管要求上。对于企业而言,了解这些差别不仅有助于其在市场上获得更好的发展机会,还能帮助其提高产品的质量和安全性。无论是生产一类、二类还是三类医疗器械,企业都应严格按照国家法规和标准进行生产管理,以确保产品的安全性和有效性,为广大的患者提供可靠的医疗保障。