随着现代医学的不断进步,医疗行业的产品种类和复杂性也在不断增加,特别是在医疗器械领域。为了确保公共健康和患者安全,国家对医疗器械实行严格的分类管理。而在这一管理框架中,医疗器械的分类无疑是影响其进入市场、审批速度及监管力度的关键因素。医疗器械的一类、二类、三类究竟有何区别?这三类产品之间的差异到底有多大呢?
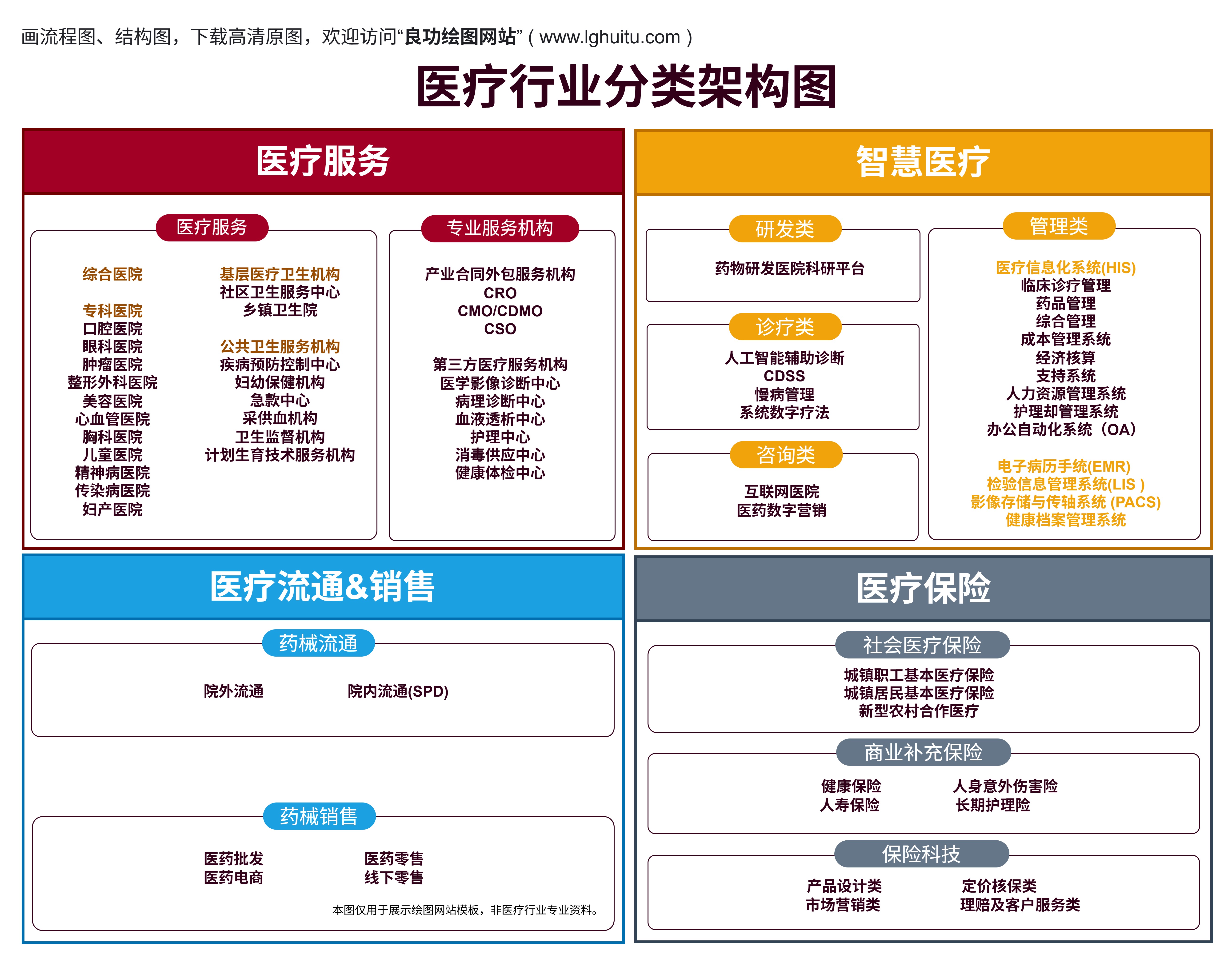
医疗器械分类制度由国家药品监督管理局(NMPA)制定,目的是根据不同医疗器械的风险程度、功能以及用途,合理地进行监管。医疗器械分类依据其风险水平的不同,分为三类:一类、二类和三类。风险低的医疗器械被归为一类,高风险的则归为三类。各类产品的审批流程、监管标准以及市场准入条件都存在明显差异,这对于生产企业来说至关重要。
一类医疗器械是风险较低的产品,通常情况下对人体的伤害较小,不会对使用者造成严重的健康危害。对于这类产品,国家的监管要求相对宽松,企业的上市程序也相对简单。例如,体温计、医用手套等都属于一类医疗器械。
一类医疗器械的管理方式通常是备案管理。也就是说,生产企业只需要向相关监管部门备案,经过产品信息的审核,确认产品符合相关安全标准后,即可在市场上销售。这一类产品一般不需要进行复杂的临床试验或长期的风险评估。
二类医疗器械的风险较一类产品更高,可能会对人体产生一定的伤害,因此需要进行更加严格的监管。对于二类医疗器械,企业在上市前需要通过国家药品监督管理局的审批,进行产品注册并提交详细的技术资料。常见的二类医疗器械包括一些诊断设备、输液器具等。

二类医疗器械的审批流程比一类要复杂,企业通常需要提供产品的安全性和有效性数据,并可能需要进行临床试验。虽然相比三类产品,其审批要求较低,但相较于一类产品,二类产品的审查会更加严格,监管措施也会更为严格。

三类医疗器械是风险最高的一类产品,这类产品通常直接影响到人体的生命健康,其安全性和有效性需要经过非常严格的验证。例如,心脏起搏器、人工关节、生命支持设备等都属于三类医疗器械。
对于三类医疗器械,企业在上市前必须提供详细的临床数据,并经过严格的审批程序。该类产品通常需要经过多轮临床试验,并且审批周期较长。为了确保产品的安全性,监管部门会对其进行全面细致的审查,并可能要求产品进行市场后续跟踪调查。由于其风险性较大,三类医疗器械的监管也最为严格。
通过以上分析,我们可以看出,一类、二类、三类医疗器械在监管标准、审批流程以及市场准入条件上有明显的差异。具体来说,三类医疗器械因其较高的风险性,在审批时需要更长时间的审查和更多的临床数据;而一类医疗器械则因为低风险,审批程序较为简便。
从产品的风险性来看,三类医疗器械的风险最高,一类医疗器械的风险最低。企业在设计和生产时,必须根据产品的风险等级采取相应的措施,以确保产品的安全性和有效性。
一类医疗器械通常只需要备案管理,审批程序相对简便。二类医疗器械则需要经过注册审批,提交详细的技术资料,审批流程较为复杂。而三类医疗器械则需要提交临床试验数据,审批周期长,且审批标准最为严格。
随着产品风险等级的增加,监管力度也逐步加大。三类医疗器械的监管要求最高,需要定期对产品进行监测和追踪。而一类医疗器械的监管相对宽松,只需定期进行产品质量检查即可。
医疗器械的分类直接影响着产品进入市场的难易程度。一类医疗器械的审批较为宽松,企业可以更快地将产品推向市场,获取利润。而三类医疗器械的审批流程则较为漫长,企业在研发和注册过程中可能面临更高的成本和风险。
医疗器械的分类还直接影响到产品的市场竞争力。一类医疗器械由于审批简单,竞争者较多,因此市场竞争激烈;而三类医疗器械则由于审批严格,进入门槛高,竞争相对较小,但产品研发的成本和周期也相对较长。
一类、二类和三类医疗器械的分类背后,不仅是监管力度的不同,还影响到企业的市场准入和产品研发策略。了解这些差异,对于医疗器械企业在产品开发和市场布局中具有重要的指导意义。每一类产品都有其独特的监管要求和市场环境,企业应根据产品的特点和市场需求选择合适的产品类型,以保证产品能够顺利进入市场并获得长期发展。
随着医疗技术的不断发展,未来的医疗器械行业可能会出现更多的新兴类别和新的监管规定,因此,企业应时刻关注行业动态,不断适应新的市场环境,提升产品的核心竞争力。