在现代医疗行业中,医疗器械的分类是保障患者安全、提升医疗质量的重要基础。根据中国国家食品药品监督管理总局(NMPA)的规定,医疗器械按照风险等级分为三类:一类、二类和三类。不同类别的医疗器械在监管标准、审批程序、市场准入条件等方面存在显著差异。了解这些分类的区别和联系,不仅能够帮助医疗行业从业者提高合规意识,也能帮助普通大众更好地理解医疗设备的安全性和重要性。

一类医疗器械是指那些对人体健康和安全危害较小、风险较低的医疗器械。这些设备通常不涉及复杂的生物学反应或长期使用所可能带来的严重副作用,因此被归类为低风险产品。根据中国的相关法规,一类医疗器械的审批程序相对简单。生产商只需进行产品备案,并且提供必要的技术资料和质量管理体系的证明即可。

常见的一类医疗器械包括一些基础的诊疗工具和设备,如体温计、血压计、手术器械等。这些设备通常不直接参与治疗过程,而更多的是用于辅助诊断或日常健康监测。
一类医疗器械的监管较为宽松,主要是因为它们的风险较低,且通常不会对患者的生命安全构成直接威胁。尽管如此,生产企业仍需确保其产品的质量符合相关标准,并且符合适用的质量管理要求,以保障使用者的安全。
与一类医疗器械相比,二类医疗器械的风险要高一些。二类医疗器械是指那些在使用过程中可能对人体健康产生中等风险的设备。它们可能涉及更复杂的技术、材料或与人体的接触较为紧密,因此需要进行更加严格的监管。
二类医疗器械的审批程序比一类更加复杂,生产商需要提交详细的产品技术资料、临床试验报告等相关证明材料,并且通过注册审批后才能上市销售。常见的二类医疗器械包括超声波诊断仪、血糖监测仪、植入性医疗器械等。这些设备通常涉及到直接的诊疗功能,甚至在某些情况下会直接影响患者的健康状况。
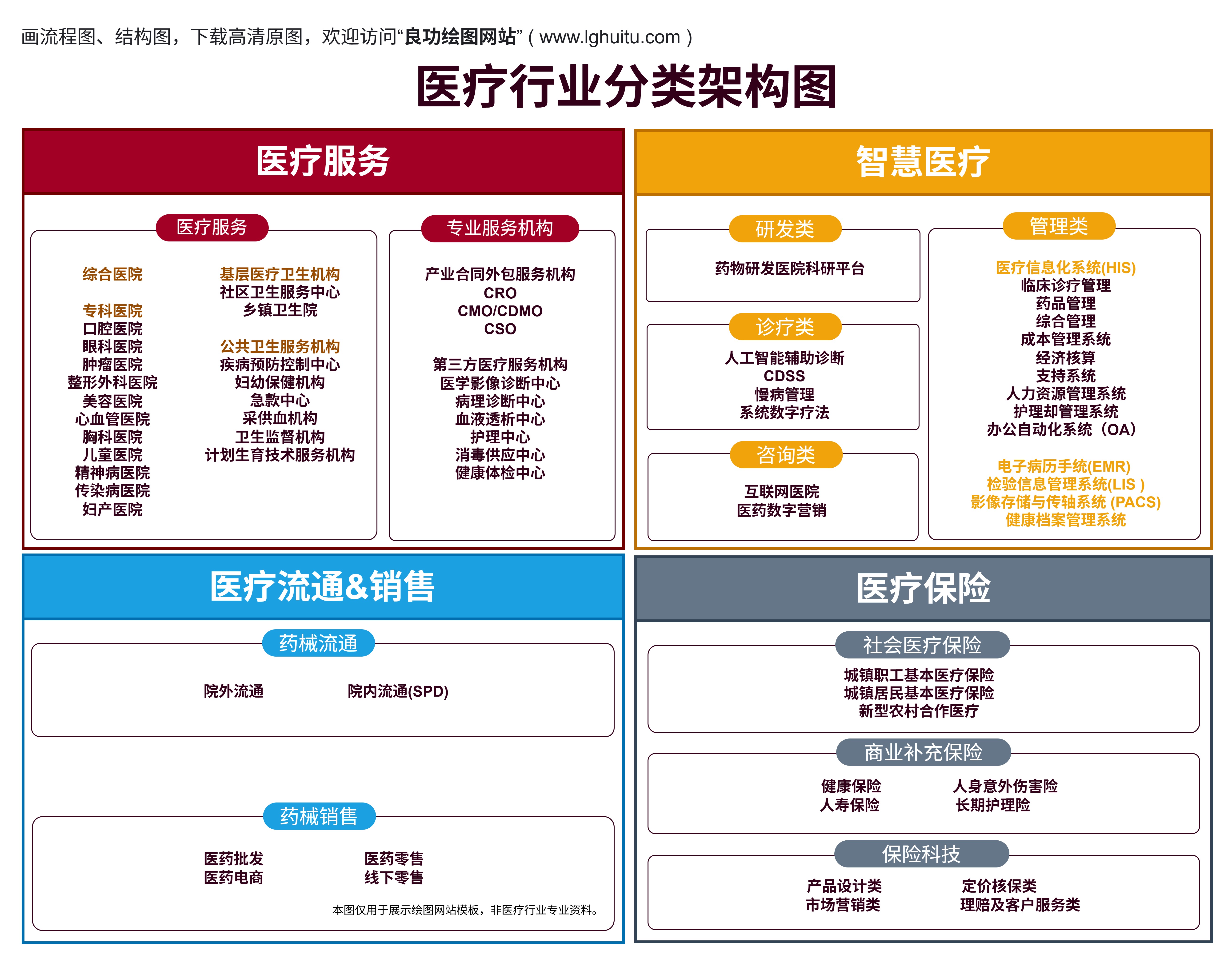
虽然二类医疗器械的风险较一类高,但它们的监管体系相对完善,且对企业的要求也较为严格。例如,生产企业需要遵守GMP(良好生产规范),并进行定期的质量检查和管理。这些措施确保了设备的安全性和有效性,减少了医疗事故的发生。
三类医疗器械是指那些对人体健康和安全具有较高风险的医疗器械。这些设备通常直接用于人体的诊疗、治疗或者对生命有重大影响。由于其潜在的高风险,三类医疗器械的审批程序最为严格,需要经过严格的临床试验、技术评估等多个环节才能获得市场准入。
三类医疗器械的典型代表包括心脏起搏器、人工关节、透析设备、放射治疗设备等。这些设备通常涉及到重大疾病的治疗,或者在手术过程中对患者的生命安全产生重要影响。因此,国家对这类设备的监管非常严格,要求企业提供详尽的临床数据、严格的质量控制和完善的售后服务体系。
为了确保这些高风险设备的安全性和有效性,三类医疗器械的审批不仅需要严格的技术评审,还需要通过较为复杂的临床试验验证。这一过程耗时长、成本高,但其核心目的就是为了确保这些设备在上市之前能够经过充分的验证,从而降低对患者的潜在风险。
一类、二类和三类医疗器械的主要区别在于它们对人体健康的风险程度不同。随着风险等级的升高,设备的监管力度也逐渐加大。从一类医疗器械的简单备案到三类医疗器械的严格审批,不同类别的医疗设备在市场准入、技术评估、临床试验等方面的要求有所不同。理解这些分类的标准,不仅帮助从业者更好地了解法规要求,也能帮助普通消费者在选择医疗设备时做出更加明智的决策。
虽然一类、二类和三类医疗器械在监管标准和审批流程上存在明显差异,但它们有一些共同点,这些共同点不仅是保障患者安全的基础,也是医疗设备分类制度的核心要素。
这三类医疗器械都必须符合国家相关的法律法规和技术标准。无论是低风险的一类医疗器械,还是高风险的三类医疗器械,它们都必须通过符合质量管理体系的认证,并保证产品的质量和性能达到相应的要求。无论哪一类医疗器械,生产企业都需要加强质量管理,确保设备的安全性和有效性。
各类医疗器械都需要接受定期的监督检查。为了确保医疗器械在市场上的安全性和可靠性,监管部门对每一类医疗器械都进行定期的抽查和检测。这是确保医疗设备在实际使用中不会对患者造成不良影响的重要保障。
不同类别的医疗器械都必须承担一定的社会责任,确保设备的使用符合伦理道德和法律规范。例如,三类医疗器械的生产企业不仅需要在技术上满足标准,还需要进行持续的临床跟踪,及时处理可能出现的任何不良反应或问题。
医疗器械分类制度在保障医疗安全、促进医疗技术发展的也为患者提供了更大的保障。从低风险的一类医疗器械到高风险的三类医疗器械,层层把关的审批制度确保了设备的安全性和有效性。在医疗行业中,了解一类、二类、三类医疗器械的区别和联系,对于行业从业者、监管部门和普通消费者来说都至关重要。
未来,随着医疗技术的不断进步,医疗器械的种类和分类标准也可能会发生变化。无论如何,确保医疗器械的安全性和可靠性始终是医疗行业最基本的要求。通过不断优化分类制度和监管机制,我们可以更好地应对医疗设备日益复杂的挑战,为公众健康保驾护航。